



New evidence on VUI-202012/01 and review of the public health risk assessment
NOVEL VARIANT INCIDENT MANAGEMENT TEAM
PUBLIC HEALTH ENGLAND WITH IMPERIAL COLLEGE, THE UNIVERSITY OF EDINBURGH, THE UNIVERSITY OF BIRMINGHAM AND THE WELLCOME SANGER INSTITUTE
Update on the submission of 11/12/20
New information:
1. Investigation of S gene target failure cases and new epidemiology data
2. Modelling on transmissibility
3. Proposed risk assessment
NERVTAG is asked to
▪ Note the new surveillance information using diagnostic PCR results
▪ Review the modelling findings
▪ Review and agree the risk assessment, specifically considering
▪ Can the variant now be said to have increased transmissibility with moderate confidence?
▪ Should impact on diagnostics be included in the risk assessment framework?
Analysis of cases with S gene target failure (SGTF) in diagnostic assays
• S-gene target failure (SGTF) in an assay commonly used in the LL has been shown through sequencing to be associated with deletions at position 69 and 70 (University of Birmingham)
• Deletion at 69/70 is found at the VUI but also in other lineages
• As of 16 Nov, of those SGTFs sequenced from the lighthouse laboratories, 87% were the VUI, rising to 97% in the latest genomic data in early December.
• These proportions will continue to be monitored
SGTF will be used as a proxy indicator for surveillance and trend analysis of the VUI from Monday 21 December (and retrospectively from 16 November onwards)
S gene target failure (provided by the Wellcome Sanger Institute)
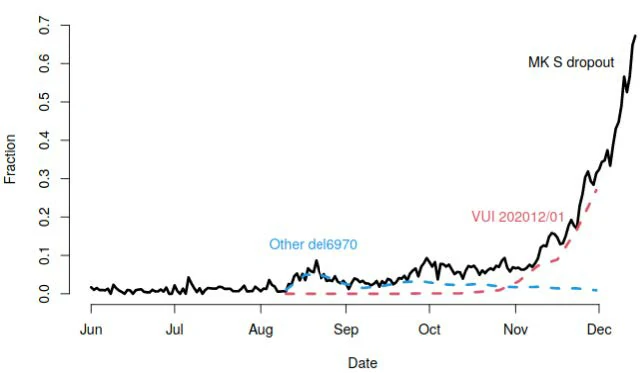
Fraction of all Milton Keynes Lighthouse Laboratory which are SGTF, and fraction of all sequenced samples which are the VUI lineage, or other lineages including the same deletion.
The proportion of cases that are SGTF at the Milton Keynes Lighthouse Laboratory has increased sharply. Of all pillar 2 samples that are sequenced, the proportion that are the VUI has shown the same trajectory, whereas other lineages with this deletion have stayed constant frequency.
Geographical distribution of S-gene target failure cases (n=67098 from 3 lighthouse laboratories)
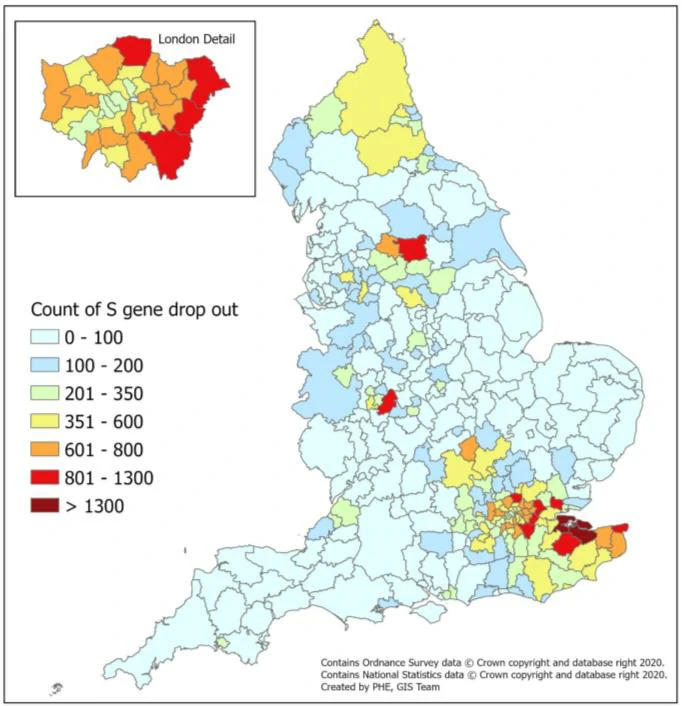
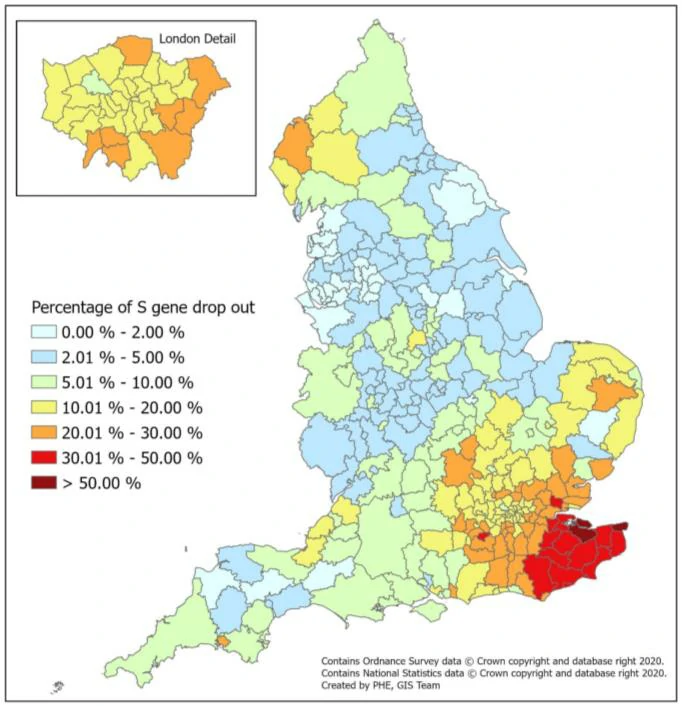
England Ad hoc (5.4) Ct Monitoring (ONS SURVEY: SHARED WITH PERMISSION)
Infectiousness within households – initial analysis
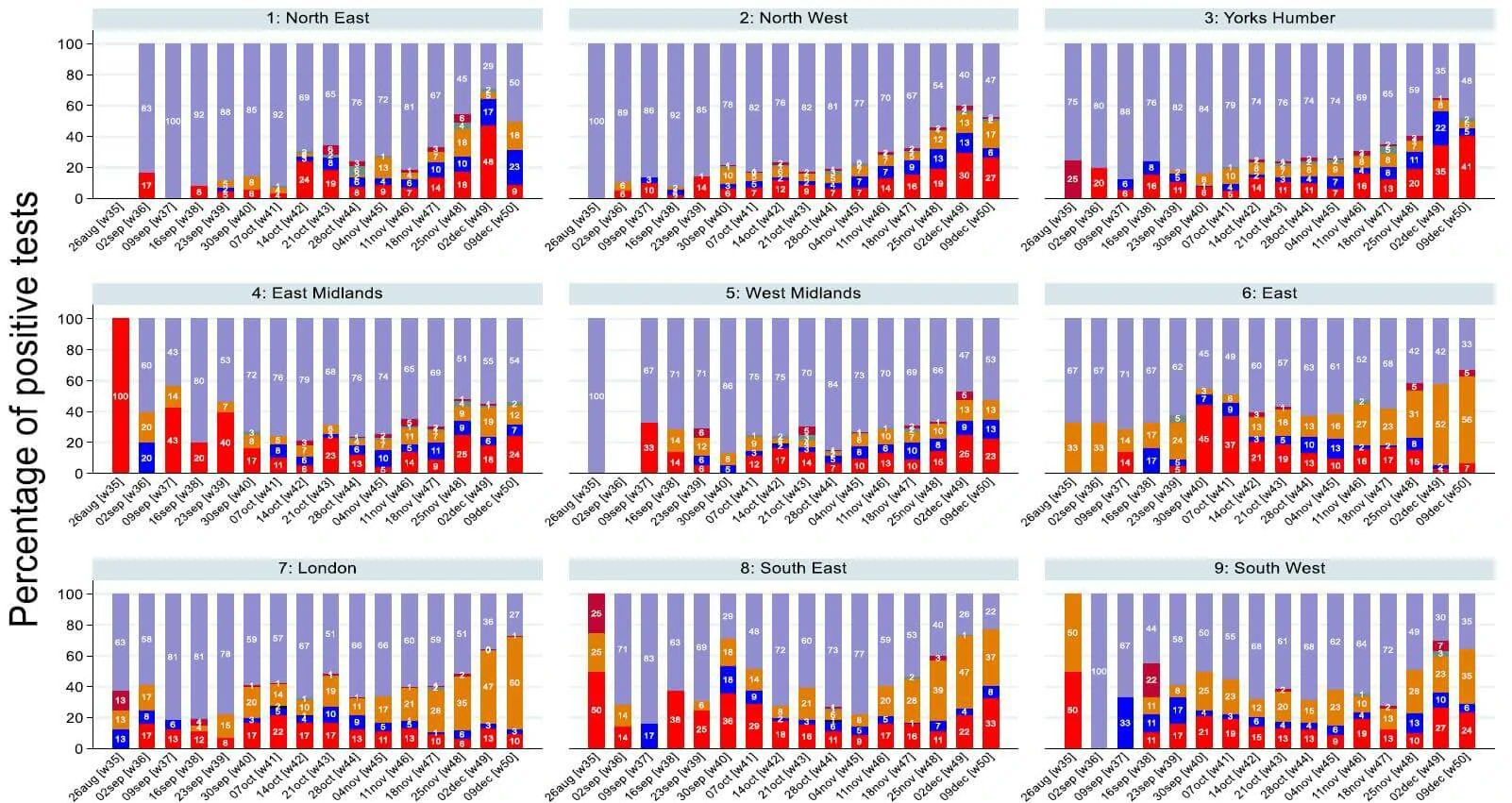
• From monitoring patterns in which genes are found to be present in the PCR test we can identify where cases of the new variant are likely to be increasing. This is because the new variant has genetic changes in the S gene, meaning that it is no longer detected by the current test.
• Samples that would previously have been positive on all three genes (purple bar on plot) are now positive only on the ORF1ab and N genes (orange bar on plot).
• Evidence suggests that the new variant is comprising an increasing proportion of positives over the last 3 weeks.
• This analysis shows that the new variant is now dominant in London and the East of England. It is also spreading to the South East and South West.
• When monitoring Ct patterns by age, there was no evidence to suggest that OR and N gene positives were more common in certain age groups than others. Whilst there was some variation by age in the whole sample, as well as in London and the East of England, this was compatible with chance.
• This analysis is presented for the whole study period up to 9 December in England regions and the Devolved Administrations on the following slide.
UK confirmed case numbers
*Cases confirmed by sequencing are likely to underrepresent total number of cases
**No new genomics data reported from COG-UK in the last 24 hours
| Total confirmed cases in the UK | New cases | % change |
| 1439 | 4 | <0.01 |
| Total presumptive cases in the UK | New presumptive cases | % change |
| 36 | 0 | 0 |
| Total confirmed cases in the UK | New cases | % change |
| 550 | 53 | 10.7 |
Proposed risk assessment
Risk Assessment
| Indicator | Risk assessment framework | Assessment (Confidence*) and rationale | |||
| Zoonotic emergence | Animal reservoir identified but no evidence of transmission from animals to humans | Sporadic transmission from animals to humans | Frequent transmission from animals to humans | NOT APPLICABLE No evidence of a zoonotic reservoir at present. |
|
| Transmissibility between humans | No demonstrated person to person transmission | Limited human case clusters | Established human to human transmission, which appears similar to wild type virus | Transmissibility appears greater than the wild type virus | RED (LOW CONFIDENCE OR MODERATE CONFIDENCE?) Preliminary modelling suggests this lineage has a high growth rate, potentially higher than other lineages co-circulating. This is biologically plausible since N501Y is in a position which could affect the receptor binding affinity of spike protein. Additional epidemiological investigations, continued surveillance and phenotypic studies are required to increase the confidence in this finding. |
| Infection severity | Evidence of less severe clinical picture or lower infection fatality than from wild type SARS-CoV-2 infections | Similar clinical picture and infection fatality to wild type SARS-CoV-2 infections OR experimental animal data suggesting potential for increased disease severity humans | More severe clinical picture or higher infection fatality than from wild type SARS- CoV-2 infections (limited to specific risk groups) | More severe clinical picture or higher infection fatality than from wild type SARS- CoV-2 infections | INSUFFICIENT INFORMATION There is no systematic data on this at present, and investigations are being urgently undertaken into deaths and hospital admissions amongst cases infected with the variant. |
| Susceptibility and immunity – natural infection | Evidence of no antigenic difference from other circulating wild type virus | Structural data suggesting antigenic difference from other circulating wild type virus | Experimental evidence of functional evasion of naturally acquired immunity | Evidence of frequent infection in humans with known prior infection with earlier virus variant. | AMBER (LOW CONFIDENCE) The N501Y variant in the spike receptor binding domain suggests that this variant may be antigenically distinct. There is no neutralisation data from polyclonal sera. The small number of possible reinfections in the variant cluster may support this but comparisons to reinfection rate in other lineages are required. Urgent neutralisation data is required. |
| Vaccines | Evidence of no structural or antigenic difference in vaccine targets | Structural data suggesting difference in vaccine target epitopes | Experimental evidence of functional evasion of vaccine derived immunity | Evidence of frequent vaccine failure or decreased effectiveness in humans | INSUFFIENT INFORMATION There is insufficient information to assess the risk of evasion of vaccine derived immunity. Urgent neutralisation data is required. |
| Drugs and therapeutics | Evidence of no structural or antigenic difference in therapeutic targets | Structural data suggesting difference in therapeutic target epitopes | Experimental evidence of reduced drug susceptibility | Evidence of frequent drug or therapeutic failure or decreased effectiveness in humans | INSUFFICIENT INFORMATION There is insufficient information to assess the risk of reduced drug susceptibility. Consideration should be given to evaluation. |
VUI-202012/01 Risk assessment part 2
Overall assessment of level and nature of risk, and level of confidence
PHE and NERVTAG consider this variant to require urgent investigation. It may be more transmissible than wild type virus, has multiple mutations, and the location of the mutations raises the possibility of antigenic change which could affect natural or vaccine derived immunity. The cluster is spreading rapidly and the lag in genomic data, combined with changes in performance in lighthouse laboratory assays, suggests it may already be widespread. This assessment is based on preliminary data. At present although experts agree, the evidence base is rapidly evolving and the current assessment has low confidence and is likely to change rapidly.
Recommendations of PHE and NERVTAG
1. Further investigations should be undertaken with urgency.
2. Surveillance should be enhanced.
3. Material should be rapidly obtained for viral culture.
4. Fitness of the mutant should be assessed in primary human airway cultures
5. Assessment of antigenicity through virus neutralisation should be made using both wild type virus and pseudovirus technical approaches.
6. Information should be sought on the genomes present in international data in GISAID with variants at position 501
7. Investigation should be undertaken to provide reassurance that Lateral Flow Devices in common use will identify this variant
8. DHSC should consider the need for enhanced control measures to limit the spread of this variant pending the availability of additional information
9. DHSC should consider the communications needed locally, nationally and internationally.
Growth in sample frequency of VUI
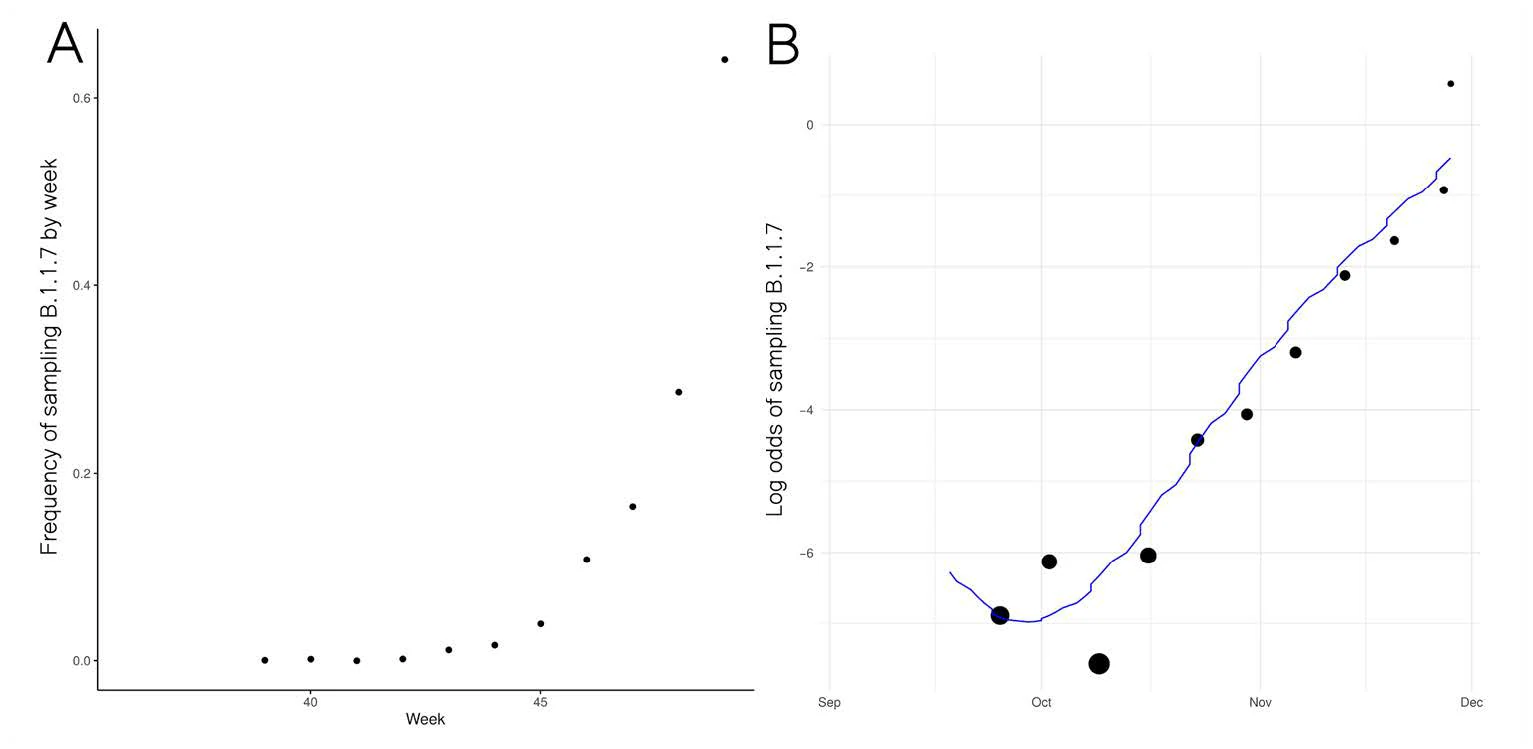
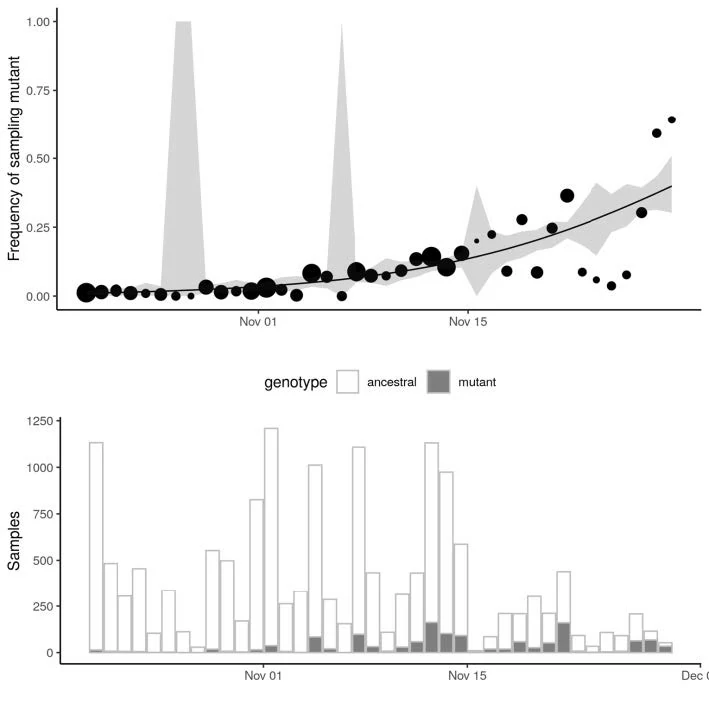
● Frequency of VUI in sequences from Pillar 2 sampling has increased exponentially since late November
● Change in frequency consistent with but not indicative of a constant selective advantage of VUI
● Logistic growth model indicates VUI grows +71% (95%CI: 67%-75%) faster per generation (6.5 days)
○ Limitations: Sample frequency is noisy & overdispersed in ways not captured by this model
Limitations: Genetic variants can achieve high frequency even if selectively neutral
Recent Example: Frequency of B.1.177 lineage in UK with A222V variant, Multiple introductions to UK in August-October 2020
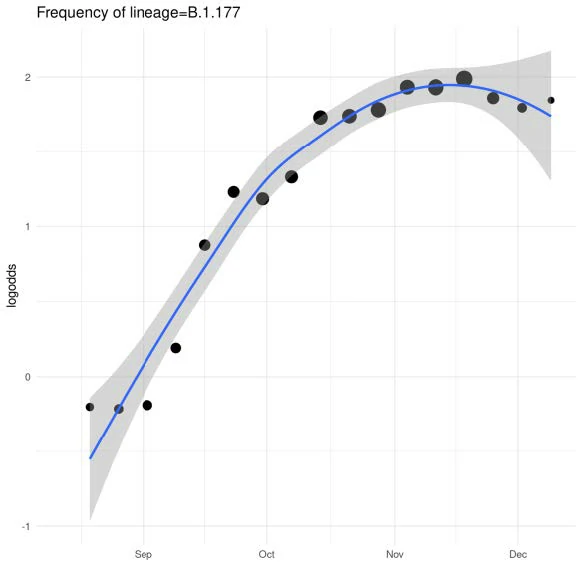
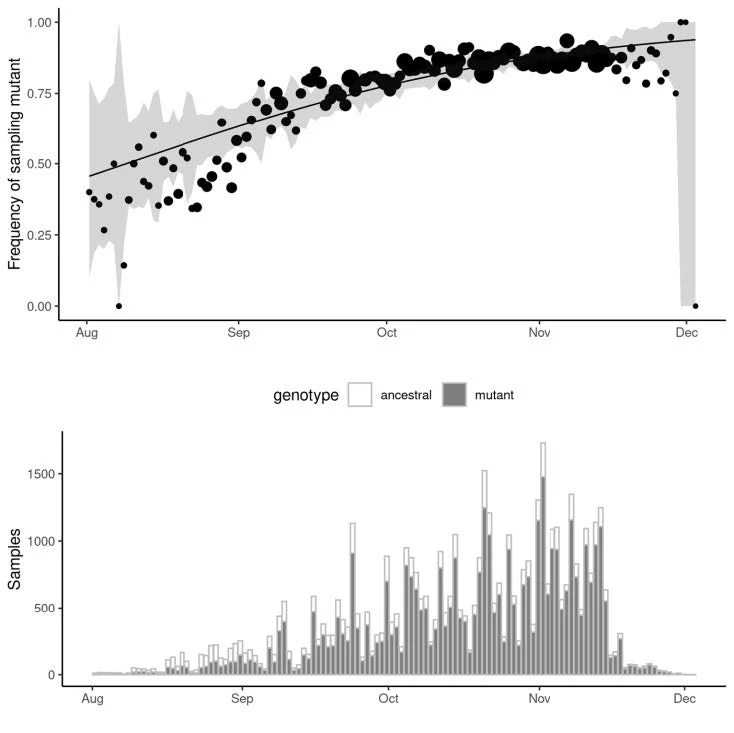
Currently >70%
frequency in UK
Initial growth fuelled by holiday travel in Europe. Growth has declined with reduced travel.
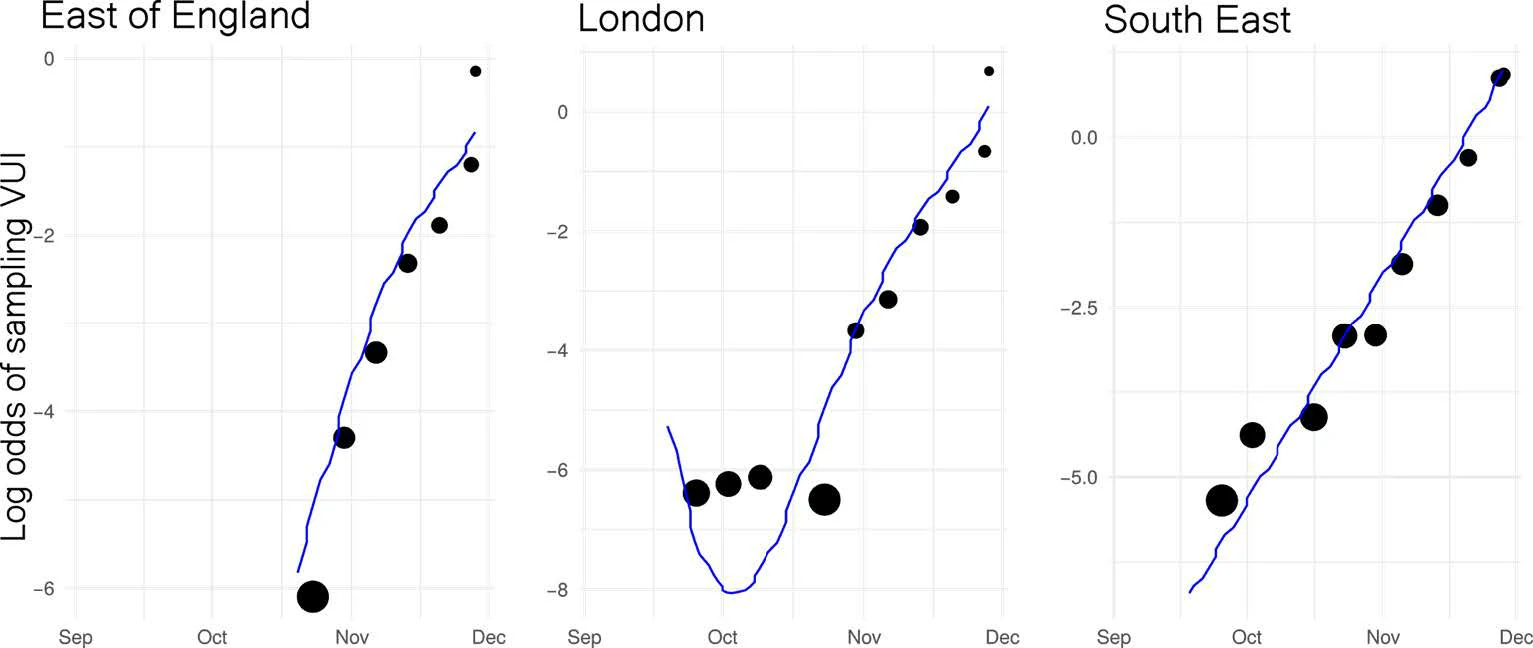
Similar rates of growth observed in different regions. Relative difference in growth rate between B.1.1.7 and other lineages:
● East of England: +72% (95%CI: 62%-82%)
● London: +86% (95%CI: 78%-94%)
● South East: +71% (95%CI: 65%-78%)
Relationship with transmission
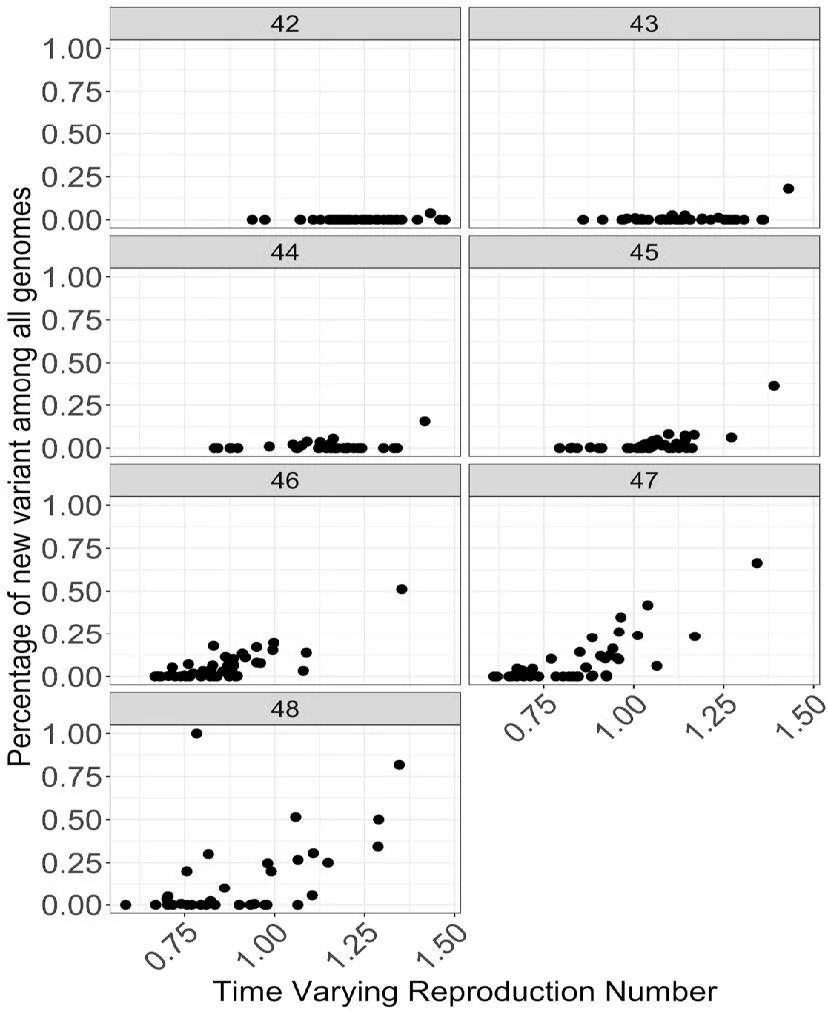
● Time varying reproduction number[1] is correlated with the increase in fraction of new variant at many places
● Figure shows relationship between fraction of new variant among all genomes plotted against the time varying reproduction number for each week. Each datapoint is an STP area.
Coalescent phylogenetic estimate
Data
1007 genomes from London and Kent sampled by Pillar 2 from 20-Sep to 30-Nov. A second analysis performed with samples up until 21-Nov to remove potential biases from lag in sequencing and non-representative sampling towards the present.
Analysis
Analysis using BEAST v1.10.4, exponential growth coalescent model, strict molecular clock.
Results – Samples from 20-Sep to 30-Nov:
Growth rate (per year): 31.96 [95% credible interval: 25.53, 38.90]Doubling time (days): 7.9 [6.5, 9.9]R: 1.57 [1.45, 1.69]
Results – Samples from 20-Sep to 21-Nov:
Growth rate (per year): 40.43 [95% credible interval: 30.66, 53.21]Doubling time (days): 6.3 [4.8, 8.3]R: 1.72 [1.55, 1.95]
Caveats
Lag in sequencing from pillar 2 results in a drop off of sequences towards the end of November –
If this is non-random then this may cause an underestimation of the growth rate.
R estimate assumes a serial interval of 6.5 days
Data
● Sequence of S:N501Y and deletion at 69-70 were used as a proxy for membership in lineage B.1.1.7
● 1451 unique pillar 2 samples collected from Sep 2nd to Nov 29 2020 across 163 local authorities areas in England
● Pillar 2 cases, deaths and new hospital admissions taken from UK dashboard
● Data aggregated by STP regions and week
Methods
● Rt for each STP per week modelled as a weekly random walk process and estimated using a semi-mechanistic Bayesian model from case and death data (Mishra, et al, medRxiv 2020.11.24.20236661; doi: https://doi.org/10.1101/2020.11.24.20236661)
● Then regress Rt for each STP against the fraction of the new variant, with categorical variables for each STP area and for each week to account for spatiotemporal effects (two variants – unweighted and weighted)
➔ Additive model : estimate the exact amount of increase or decrease in Rt by using Rt as response in the linear model
➔ Multiplicative model : estimate the relative increase or decrease in Rt by using log(Rt) as the response variable in the linear model
Results
● Additive model (unweighted): increase in Rt of 0.39 [0.24-0.55] ➔ For example, under the additive assumption, an area with an Rt of 0.8 without the new variant would have an Rt of 1.19 [1.04-1.35] if only N501Y was present
● Additive model (weighted): increase in Rt of 0.93 [0.73-1.13]● Multiplicative model: relative increase in Rt of 48% [27%-74%] ➔ For example, under the multiplicative assumption, an area with an Rt of 0.8 without the new variant would have an Rt of 1.18 [1.02-1.40] if only N501Y was present
Limitations and assumptions
● Frequency may be underestimated from genomic data
● Confidence intervals assume independence of the observations, homoscedasticity and normality of the observations
● Spatial correlation has not be taken into consideration
● Population is considered homogeneous and all age bands are considered equally
● No causal relationship established. Only associative effects are estimated
