



AAV vector system, AAV production, transduction and AAV gene therapy
SOCAIL MEDIA

Adeno-associated virus (AAV vector) -Introduction
Landscape, protocol and guidelines of Adeno-associated virus (AAV) vector system, AAV production,AAV transduction and AAV gene therapy are described below. The detail information is mentioned about AAV vector plasmids cloning, AAV packaging, purification, AAV capsid evolution and insight of AAV gene therapy.
Table of Contents
Adeno-associated virus (AAV vector) and AAV gene therapy-Knowledge Base
1. What is Adeno-associated virus (AAV) vector?
Adeno-associated virus (AAV) is a small-single strand DNA virus, member of human parvovirus, originally described in the 1960s by Bob Atchison at Pittsburgh and Wallace Rowe at NIH as a contaminant in preparation of adenovirus [1,2]. Shortly after its discovery, AAV was then isolated from humans, but serological studies suggested that AAV itself did not cause any disease despite being present in people infected with helper viruses such as adenovirus or herpes virus [3]. With the development of recombinant AAV vectors (rAAV), the adenovirus helper genes required for AAV replication have been identified, which can be cloned into plasmids for AAV production to liberate the need of helper virus. Currently, AAV production adopts the 3-plasmid co-transfection system (AAV plasmid with gene of interest, AAV packaging plasmid AAV-RC (AAV replication and AAV capsid) and pHelper (AAV helper plasmid)) in AAV-293 cells, which significantly simplifies the AAV purification process.
2. AAV Gene Therapy Review
AAV has been proved as the most excellent gene therapy vector. To date, more than 204 clinical trials have been carried out using AAV vectors for gene delivery [4], and promising gene therapy outcomes have been achieved from Phase 1, Phase 2 and Phase 3 trials for a great number of diseases (Table 1), including lipoprotein lipase deficiency (LPLD) [5], spinal muscular atrophy (SMA) [6], retinal dystrophy [7,8], cystic fibrosis [9,10], Duchenne Muscular Dystrophy [11], Hemophilia [12], congestive heart failure [13], Parkinson’s disease [14] and Rheumatoid Arthritis [15,16]. Among them, Luxturna from Spark has been approved by FDA to treat patients with retinal dystrophy, AVXS-101 by AveXis has shown outstanding therapeutic effects in patients with spinal muscular atrophy, while AAVs with micro-dystrophin and coagulation factor IX have displayed great potential in the treatment of Duchenne Muscular Dystrophy and Hemophilia respectively.
a)Luxturna for treatment of retinal dystrophy
The FDA has approved LUXTURNA™ (voretigene neparvovec-rzyl), a one-time gene therapy product designed for the treatment of the LCA ( Leber’s congenital amaurosis) patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy in December 2017. LUXTURNA is the first and only pharmacologic treatment for an inherited retinal disease (IRD) and the first AAV vector gene therapy for a genetic disease approved in the U.S. LUXTURNA uses AAV2 to carry out functional copies of the RPE65 gene delivery to the retinal pigment epithelial (RPE) cells to compensate for the RPE65 mutation [17]. With the functional RPE65 gene delivery, retinal pigment epithelial cells begin producing the RPE65 protein [18], letting 11-cis-retinal (a critical visual pigment component) regenerate to restore the visual cycle [4,19]. To date, Phase 1 studies have shown potential benefits of gene replacement in RPE65-mediated inherited retinal dystrophy, and the Phase 3 studies also demonstrate great efficacy and safety of Luxturna gene therapy in participants inherited retinal dystrophy [20].
| Indication | Gene | AAV serotype | Route of AAV delivery | Phase | Sponsor |
| Lipoprotein lipase deficiency | LPL | AAV1 | Intramuscular | II–Ⅲ | Amsterdam Molecular Therapeutics |
| LPL | AAV1 | Intramuscular | I | UniQure Biopharma B.V. | |
| Spinal muscular atrophy 1 | SMN | AAV9 | Intravenous | I | AveXis |
| SMN | AAV9 | Intravenous | Ⅲ | AveXis | |
| SMN | AAV9 | Intravenous | Ⅲ | AveXis | |
| Spinal muscular atrophy | SMN | AAV9 | Intravenous | I | AveXis |
| SMN | AAV9 | Intravenous | Ⅲ | AveXis | |
| SMN | AAV9 | Intravenous | Ⅲ | AveXis | |
| Retinal dystrophy | RPE65 | AAV2 | Subretinal | I–II | University College, London |
| PDE6B | AAV5 | Subretinal | I–II | Horama S.A | |
| Cystic fibrosis | CFTR | AAV5 | Lung, via aerosol | I | NIDDK |
| Hemophilia A | HLP-FVIII-V3 | AAV8 | Intravenous | I | University College, London |
| Hemophilia B | FIX | AAV2 | Intravenous | I | Spark Therapeutics |
| FIX16 | AAVrh10 | Intravenous | I–II | Ultragenyx Pharmaceutical Inc | |
| Arthritis | TNFR:Fc | AAV2 | Intraarticular | I | Targeted Genetics Corporation |
| TNFR:Fc | AAV2 | Intraarticular | I–II | Targeted Genetics Corporation | |
| hIFN-b | AAV5 | Intraarticular | I | Arthrogen | |
| hIFN-b | AAV5 | Intraarticular | I | Arthrogen | |
| Congestive heart failure | SERCA2a | AAV1 | Intracoronary | I–II | Celladon Corporation |
| SERCA2a | AAV1 | Intracoronary | II | Celladon Corporation | |
| SERCA2a | AAV1 | Intracoronary | II | Imperial College London | |
| SERCA2a | AAV1 | Intracoronary | II | Assistance Publique – Hôpitaux de Paris | |
| SERCA2a | AAV1 | Intracoronary | I–II | Celladon Corporation | |
| Parkinson’s disease | GAD | AAV2 | Surgical infusion | I | Neurologix, Inc. |
| GAD | AAV2 | Intrastriatal | I | Genzyme, a Sanofi Company | |
| NTN | AAV2 | Intrastriatal | I | Genzyme | |
| NTN | AAV2 | Bilateral Intraputaminal (IPu) | II | Genzyme | |
| NTN | AAV2 | Bilateral Intraputaminal and Intranigral | I–II | Sangamo Therapeutics | |
| Alzheimer’s disease | NGF | AAV5 | Bilateral, stereotactic | I | Ceregene |
| APOE2 | AAVrh.10 | Intracisternal | I | Weill Medical College of Cornell University |
b)AVXS-101 for treatment of spinal muscular atrophy (SMA)
AVXS-101 is being developed by AveXis (Owned by Novartis) to carry out gene delivery of a functional copy of the SMN1 gene by using AAV9 vector to motor neuron cells in SMA patients [21]. The SMN1 transgene in AVXS-101 contains double-stranded DNA, meaning it consists of the genetic instructions and takes the same form as natural genes, so that it can be activated more quickly to produce continuous and sustainable SMN protein, resulting in more efficient gene therapy in vivo. The safety, tolerability and impressive improvements in motor function of AVXS-101 in all patients with SMA Type 1 has been guaranteed during Phase 1 clinical trials [51]. Based on the preliminary top-line results, the FDA has granted AVXS-101 Orphan Drug Designation for the treatment of all types of SMA and Breakthrough Therapy Designation, as well as Fast Track Designation, for the treatment of SMA Type 1. Likewise, the European Medicines Agency (EMA) also granted AveXis access into its PRIority Medicines (PRIME) program for AVXS-101 for the treatment of SMA Type 1 in January 2017. Then in December 2017, AveXis also announced the start of an open-label, dose-comparison, multi-center Phase 1 trial, also known as STRONG, to evaluate the safety, optimal dosing and proof of concept for efficacy of AVXS-101 gene therapy in two distinct age groups of patients with SMA Type II. Nowadays, AveXis is running a Phase 3 trial, called STR1VE (NCT03306277) in infants with SMA type 1 to further understanding of both the safety of AVXS-101 gene therapy, and how well AVXS-101 may work in SMA patients, which may bring new hope for pediatric and adult patients with other types of motor neuron diseases.
c)AAV with micro-dystrophin for treatment of Duchenne Muscular Dystrophy
Duchenne muscular dystrophy (DMD), resulting from dystrophin gene mutation, is a severe genetic disease resulting in body-wide muscle degeneration and necrosis in boys and young men [22]. In gene therapy theory, replacing or correcting the mutated dystrophin gene with a functional one would cure this disease [23]. However, the enormous size of dystrophin cDNA and the distribution of muscle throughout the body presented great challenges to this arduous task. To address these obstacles, researchers have developed the highly abbreviated micro-dystrophin gene and designed systemic gene delivery with AAV2.5, which was a translational optimized AAV variant, inducing stronger transduction in skeletal muscles than AAV2 and displaying lower crossreactivity to AAV2 neutralizing antibodies [11]. To date, preclinical data suggests that intravascular AAV micro-dystrophin delivery can significantly ameliorate muscle pathology, enhance muscle force, and attenuate dystrophic cardiomyopathy in animals, such as mice and canines [24,25]. Phase 1 clinical trials of AAV2.5 in DMD patients have shown that rationally designed AAV vector (AAV2.5) was safe and well-tolerated, setting the foundation of customizing AAV vectors best suiting the clinical objectives [11]. Meanwhile, more clinical trials are required to estimate the clinical efficacy of AAV2.5 with micro-dystrophin gene, and the findings display great prospects of body-wide DMD therapy with a synthetic micro-dystrophin AAV vector [26].
d)AAV with coagulation factor IX for treatment of Hemophilia B
Hemophilia B (HB) is an X-linked disease, caused by a deficiency in functional coagulation factor IX protein (FIX) [27]. Currently, preclinical data suggests AAV vector-mediated gene transfer of coagulation factor IX to the skeletal muscle or liver have shown sustained correction of hemophilia B in mice and dogs [28,29]. The two initial phase I/II clinical trials with FIX cDNA gene delivery by AAV vectors for treatment of hemophilia B displayed no serious adverse effects, but demonstrated contrary results, no therapeutic level of FIX in muscle trial while some therapeutic effects closely with that of hemophilic dogs [30]. To date, gene therapy for Hemophilia B via systemic administration of AAV vectors containing an optimized coagulation factor IX construct has achieved considerable progress, ameliorating the bleeding phenotype numerous patients [31]. However, challenges remain for sustained gene therapy in patients, and efforts are still required to improve long term expression of factor IX, via optimizations to the AAV vector, transgene cassette and correction strategy.
3. Advantages and drawbacks of AAV vector-mediated gene transfer
a) Advantages of AAV-mediated gene transfer
AAV has been developed into a very attractive candidate for creating viral vectors for gene therapy and the creation of isogenic human disease models due to various advantages. 1) Superior biosafety rating. The wild type AAV has not currently been known to cause disease in vivo, and further security of recombinant AAV gene delivery in vivo is ensured after removal of most AAV genome elements. 2) Low immunogenicity. AAV causes a very mild immune response in vivo, lending further support to its apparent lack of pathogenicity during gene delivery. 3) Broad range of infectivity. AAV can infect both dividing and quiescent cells in vivo, allowing gene delivery to a highly diverse range of cell types. 4) Stable expression. Long term gene delivery in vivo can be mediated by AAV.
b) Drawbacks of AAV-mediated gene transfer
Although adenovirus benefits a great deal of disease therapies, it does present some drawbacks.
1) The major drawback is its limited cloning capacity (less than 4.7kb) of the vector, which restricts its use in gene delivery of large genes.(Table 3) [32]. 2) Generation of neutralizing antibodies against AAV in the Non-Human Primates (NHP) and human, may attenuate the cure effect of AAV-mediated gene therapy [33].
| Comparison | Retrovirus | Lentivirus | Adenovirus | AAV |
| Genome | ss RNA | ss RNA | ds DNA | ss DNA |
| Integration | Yes | Yes | No | No |
| Packaging Capacity | 3kb | 4kb | 5.5kb | 2kb |
| Time to peak expression | 72h | 72h | 36h-72h | Cell: 7 days; Animals: 2 weeks |
| Sustainable time | About 3 weeks | Stable expression | Transient expression | > 6 months |
| Cell Type | Most Dividing | Most Dividing/Non-Dividing Cells | Most Dividing/Non-Dividing Cells | Most Dividing/Non-Dividing Cells |
| Titer | 10^7 TU/ml | 10^8 TU/ml | 10^11 PFU/ml | 10^12 vg/ml |
| Animal experiment | Suitable | Low efficiency | Lowest efficiency | Most suitable |
| Immune Response | High | Medium | Medium | Ultra-low |
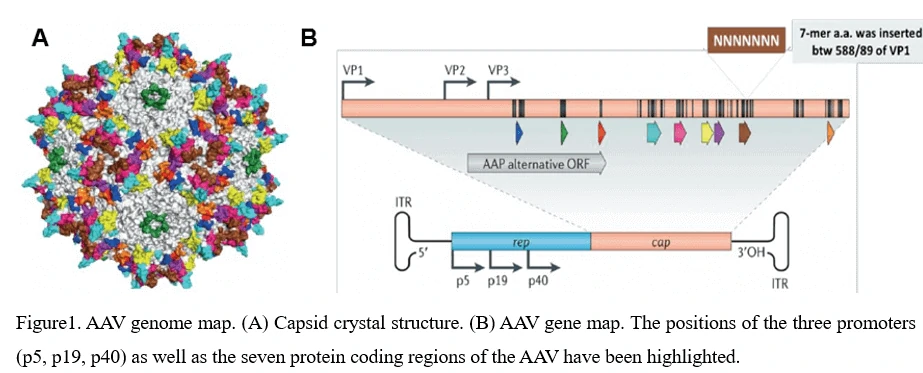
4. AAV Genome Structure (ITR,Rep,Cap,VP1,VP2,VP3) and AAV virus assembly
The AAV shell is approximately 25nm in diameter and encapsidates a single-stranded DNA genome of 4.7 kilobases. The genome consists of two large open reading frames (ORFs) flanked by 145bp inverted terminal repeats (ITR), which are the only cis-acting elements required for AAV genome replication and AAV packaging. The left ORF encodes four replication proteins, Rep40, Rep52, Rep68, and Rep78, in charge of site-specific integration, as well as regulation of AAV capsid formation initiation within the AAV genome, while the right ORF encodes the viral structural proteins, VP1, VP2, and VP3, which interact together to assemble into icosahedral virion shells comprising 60 subunits each (Figure1).
5. Life Cycle of AAV Virus-From Infection To Regeneration
AAV transduces cells through several stages: (1) viral binding to cell surface receptor/coreceptor, (2) endocytosis of the virus, (3) intracellular trafficking of the virus through the endosomal compartment, (4) endosomal escape of the virus, (5) intracellular trafficking of the virus to the nucleus and nuclear import, (6) virion uncoating, (7) viral genome conversion from a single-stranded to a double-stranded genome capable of expressing an encoded gene [34-36]. Since AAV can’t encode polymerases, AAV is dependent upon cellular polymerase activity to replicate its own genome [37]. The presence of a helper virus such as adenovirus is indispensable for wild-type AAV to facilitate gene expression and replication. (Figure 2A) Without helper virus, expression of Rep68/Rep78 would be restricted owing to Ying Yang 1 (YY1) repression of the P5 promoter, leading to inhibition of AAV genome replication and gene expression, and initiation of AAV chromosome integration (Figure 2B) [38]. AAV establishes latency by undergoing specifically integration into a genome site, termed as the adeno-associated virus integration site 1 (AAVS1), a 4kb region on chromosome 19 (q13.4).
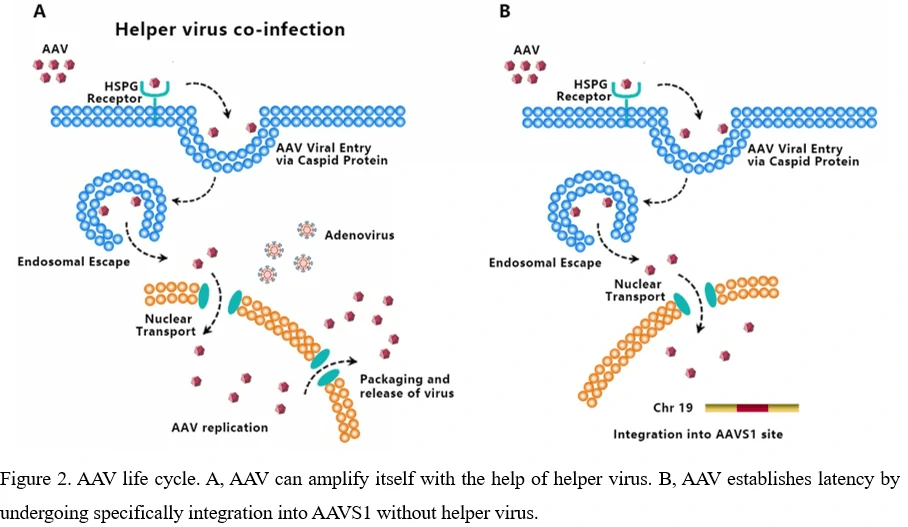
So far, no known disease has been demonstrated to be related to AAV though AAV has been detected in many tissues of animal species. Serological studies show that most people are exposed to wild-type AAV during childhood and adolescence and prevalence rates for antibody titers exceed 60% among adults, while no detrimental effects or lytic infections have been observed [39,40].
6. AAV serotypes and AAV Tissue-specific Tropism
Over the past decades, numerous AAV serotypes have been identified with variable tropism. To date, 12 AAV serotypes and over 100 AAV variants have been isolated from adenovirus stocks or human/nonhuman primate tissues. Among them, AAV2, AAV3, AAV5, AAV6 were discovered in human cells, while AAV1, AAV4, AAV7, AAV8, AAV9, AAV10 (AAVrh10), AAV11, AAV12 in nonhuman primate samples [41]. Genome divergence among different serotypes is most concentrated on hypervariable regions (HVRs) of virus capsid, which might determine their tissue tropism[42]. Besides virus capsid, tissue tropisms of AAV vectors are also influenced by cell surface receptors, cellular uptake, intracellular processing, nuclear delivery of the vector genome, uncoating, and second-strand DNA conversion [3]. To effectively enable the transduction of tissues or limit AAV tropism to specific tissues in vivo, researchers have developed a number of mosaic AAV vectors or chimeric AAV vectors by engineering custom-designed AAV capsids to carry out gene therapy [43,44].
a)AAV Serotype 2 (AAV2)
AAV Serotype 2 (AAV2) is the first isolate of AAVs and also the best studied AAV serotype, which stands for natural tropism towards skeletal muscles [45], neurons [46], vascular smooth muscle cells [47], and hepatocytes [48]. AAV2 possesses three receptors, among which heparan sulfate proteoglycan (HSPG) functions as a primary receptor to scavenge AAV particles and impair the infection efficiency [49], while aVβ5 integrin and fibroblast growth factor receptor 1 (FGFR-1) enable AAV to enter the cell by receptor-mediated endocytosis as co-receptors [50-52]. In possession of great gene therapy potentials, AAV2 has been used for cancer treatments, such as ovarian carcinoma (OvCa) without any effect on healthy cells currently [53].
b)AAV Serotype 8 (AAV8) and AAV Serotype 9 (AAV9)
In addition to AAV2, the most widely used AAV serotypes are AAV Serotype 8 (AAV8) and AAV Serotype 9 (AAV9). AAV8 is a robust vector for gene delivery to liver and a promising research tool for delivering genes to various target organs with higher efficiency than most other serotypes. As demonstrated, efficient transduction of liver, skeletal muscle and heart can be detected after systemic injection of AAV8 [54]. More importantly, a single tail vein injection of AAV8 was as efficient as portal vein injection at any dose and a high dose of AAV8 transduced skeletal muscle throughout the body, including the diaphragm, the entire cardiac muscle, and the pancreas, smooth muscle, and brain at substantial levels [55]. Compared with AAV2, AAV9 displays a similar profile with widely disseminated transduction, just with much more efficient [56]. Additionally, the ability of AAV9 to bypass the blood-brain barrier (BBB) enables intravascular administration as a non-invasive delivery route to achieve widespread CNS gene expression. What’s more, AAV9 intravenous gene delivery has already shown promising results for several diseases in animal models, including lysosomal storage disorders and motor neuron diseases, opening the way to the first clinical trial in the field [57].
c)Other AAV serotypes
Today, 12 AAV serotypes have been designed as gene delivery vectors. It might be difficult to interpret AAV serotype tissue tropism due to interstudy variations in vector titers and doses, promoters, and transgenes. A general hierarchy of transduction efficiency in major tissues and cell types has been established at table 1 and table 2. For instance, AAV2 can moderately transduce several tissue types, including central nervous system, liver, muscle, and lung. Similarly, within the CNS, AAV1 and AAV5 show higher transduction frequencies than AAV2 in all injected regions [58,59], while AAV4 just appears to transduce specific cell types, such as the astrocytes and ependyma in the subventricular zone [60]. In skeletal muscle cells, AAV1, AAV6 and AAV7 are reported to present a very high transduction rate [56,61-63], while AAV8 exhibits transduction ability with super efficiency not only in hepatocytes but also in other organs [44,61,64]. In addition to skeletal muscle cells, AAV6 has been also reported to mediate more efficient transduction of airway epithelial cells in mouse lungs compared to that of AAV2 [64], which may supply AAV6 with more significant advantages over AAV2 for gene therapy of lung diseases like cystic fibrosis considering the lower immunogenicity of AAV6 than of AAV2 [44,61].
| AAV Serotype | Tissue tropism | |||||||
| CNS | Retina | Lung | Liver | Pancreas | Kidney | Heart | Muscle | |
| AAV1 | √ | √ | √ | √ | √ | |||
| AAV2 | √ | √ | √ | |||||
| AAV3 | √ | √ | √ | √ | ||||
| AAV4 | √ | √ | √ | |||||
| AAV5 | √ | √ | √ | √ | ||||
| AAV6 | √ | √ | √ | √ | √ | |||
| AAV7 | √ | √ | ||||||
| AAV8 | √ | √ | √ | √ | ||||
| AAV9 | √ | √ | √ | √ | √ | |||
| AAV-DJ | √ | √ | √ | √ | ||||
| AAV-DJ/8 | √ | √ | √ | |||||
| AAV-Rh10 | √ | √ | √ | √ | √ | |||
| AAV-retro | √ | √ | √ | |||||
| AAV-PHP.B | √ | √ | √ | |||||
| AAV8-PHP.eB | √ | √ | ||||||
| AAV-PHP.S | √ | √ | √ | |||||
| Cell Line | AAV1 | AAV2 | AAV3 | AAV4 | AAV5 | AAV6 | AAV8 | AAV9 | AAV-DJ | AAV-DJ/8 |
| Huh-7 | 13 | 100 | 2.5 | 0 | 0.1 | 10 | 0.7 | 0 | 500 | 0.2 |
| HEK293 | 25 | 100 | 2.5 | 0.1 | 0.1 | 5 | 0.7 | 0.1 | 500 | 0.3 |
| Hela | 3 | 100 | 2 | 0.1 | 6.7 | 1 | 0.2 | 0.1 | 667 | 0.2 |
| HepG2 | 3 | 100 | 16.7 | 0.3 | 1.7 | 5 | 0.3 | ND | 1250 | 0.5 |
| Hep1A | 20 | 100 | 0.2 | 1 | 0.1 | 1 | 0.2 | 0 | 400 | 0.1 |
| 911 | 17 | 100 | 11 | 0.2 | 0.1 | 17 | 0.1 | ND | 500 | 0 |
| CHO | 100 | 100 | 14 | 1.4 | 333 | 50 | 10 | 1 | 25000 | 5 |
| COS | 33 | 100 | 33 | 3.3 | 5 | 14 | 2 | 0.5 | 500 | 0.3 |
| MeWo | 10 | 100 | 20 | 0.3 | 6.7 | 10 | 1 | 0.2 | 2857 | 1 |
| NIH3T3 | 10 | 100 | 2.9 | 2.9 | 0.3 | 10 | 0.3 | ND | 500 | 0.1 |
| A549 | 14 | 100 | 20 | ND | 0.5 | 10 | 0.5 | 0.1 | 1000 | 0.1 |
| HT1180 | 20 | 100 | 10 | 0.1 | 0.3 | 33 | 0.5 | 0.1 | 333 | 0.2 |
| Monocytes | 1111 | 100 | ND | ND | 125 | 1429 | ND | ND | 100 | ND |
| Immature DC | 2500 | 100 | ND | ND | 222 | 2857 | ND | ND | 200 | ND |
| Mature DC | 2222 | 100 | ND | ND | 333 | 3333 | ND | ND | 100 | ND |
7. AAV Vector System (AAV Vector System)
a)Introduction of rAAV vector system
Though wild-type AAV is not associated with human disease, it is naturally defective and requiring helper adenovirus or herpes simplex virus (HSV) coinfection for AAV replication, so recombinant AAV (rAAV) has been an attractive vector for gene therapy. In 1984, rAAV was used for the first time by replacing the viral genome with a transgene, which was then transfected into cells that were infected with adenovirus for production of rAAV.
Traditionally, rAAV vectors used in clinical trials were prepared with a plasmid containing the therapeutic gene flanked by AAV-inverted terminal repeats (ITRs), co-transfected with AAV packaging plasmid AAV-RC (AAV replication and AAV capsid) and pHelper (AAV helper plasmid). The adenovirus helper factors, such as E1A, E1B, E2A, E4ORF6 and VA RNAs, would be provided by the third helper plasmid. Given that HEK293 cells, a commonly used AAV production cells, already contain the E1A/E1b gene, only E2A, E4ORF6 and VA RNAs need to be provided by the helper plasmid.
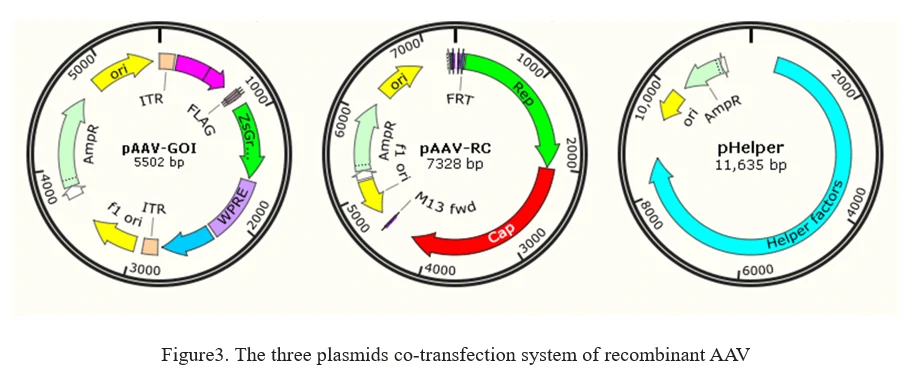
b)Helper-free rAAV system
The current method of the rAAV production is based on the adenovirus-free transient transfection of all elements that are required for AAV production in host cells, such as HEK293 cells. It involves the co-transfection of 3 plasmids into AAV production cells as shown in Figure 3.
1. AAV-GOI: an AAV ITR-containing plasmid carrying the gene of interest (GOI);
2. AAV-RC: an AAV serotype plasmid that carries Rep and Capsid genes;
3. pHelper: an AAV helper plasmid that provides the helper genes isolated from adenovirus.
8. AAV engineering (AAV capsid evolution)
Although more than 100 AAV serotype variants have been found in nature, significant progress has been made for rAAV production, the transduction efficiency and specificity for some tissues and cell types in animals and primates is low. The non-targeted delivery by AAV requires high doses and potentially resulting in toxicities, problematic manufacturing burdens and suboptimal efficacy. Therefore, an important area in the development of AAV as a vector concerns the engineering of altered cell tropisms to increase rAAV mediated gene delivery efficiency and specificity. Among various AAV serotypes, the variations in transducing efficiencies could be caused by differences in their respective receptor contents on target cells.
A great number of strategies for improving AAV delivery efficiency and specificity have been developed. Methodologically, the methods to improve AAV tropism and specificity can be divided into two categories. One is the assembly and recombination of proteins between different viruses, which can produce hybrid rAAVs. The other is to recombine, redesign, or introduce random mutations into the capsid protein of AAV by different methods to artificially increase the variance of AAV serotypes, and then screen the appropriate AAV serotypes.
a)Hybrid rAAV Vectors
The commonly used approach to generate hybrid rAAV vectors is transcapsidation, which is a process involving the packaging of the ITR from one AAV serotype into the capsid of another serotype, which may determine the tissue tropism of hybrids. For instance, if it is rAAV2/5 that is to be produced based on the helper-free system discussed above, the AAV packaging plasmid AAV-RC should be pRep2/Cap5, that is, the Rep gene is derived from AAV2, while the Capsid gene from AAV5. The packaging scheme is the same as the above-mentioned approach for AAV2 production. The resulting rAAV is named rAAV2/5, in which the genome is based on recombinant AAV2, while the capsid based on AAV5. The tissue tropism displayed by this rAAV2/5 hybrid virus is assumed to be the same with AAV5.
b)AAV revolution
There are two main ways to evolve AAV, one is rational design, the other is direct evolution (Figure4).
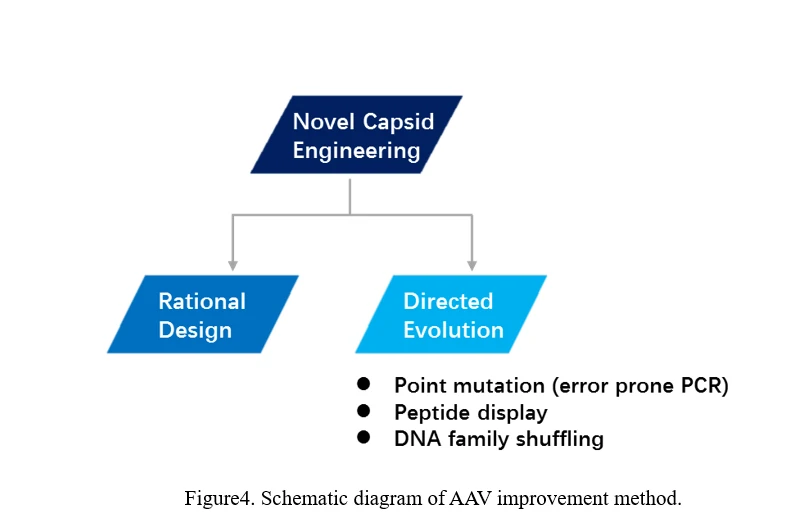
Rational Design
Some common methods of rational design AAV capsid, based on knowledge about delivery mechanisms coupled with AAV structural analysis [65], are listed as follows.
(1) Phosphorylation of tyrosine residues in AAV capsid results in ubiquitylation and promotes proteasomal degradation of AAV virions, leading to the creation of vectors in which tyrosines were mutated to phenylalanines by site-directed mutagenesis [66].
(2) Analysis of potential docking sites for a murine IgG2a antibody with the AAV2 surface to determine sterically accessible candidate positions, which were then subjected to extensive site-directed mutagenesis to develop variants with reduced neutralization by antibodies in vitro [67].
(3) Incorporation of high-affinity ligands into the AAV capsid can confer binding to alternative cell surface receptors and thereby restrict or redirect viral tropism [68].
AAV Directed Evolution
The conventional process of directed evolution is generating a library by error-prone PCR, peptide display or DNA family shuffling at firs. Then, this AAV viral library was packaged as a pool and purified, and selected under certain pressure in vitro or in vivo. After infection of the target cell or tissue, the viruses would recover from adenovirus-mediated replication or PCR amplification. Three or more rounds selection later, the highly infectious and specific AAV capsid species would be enriched (Figure 5) [69].
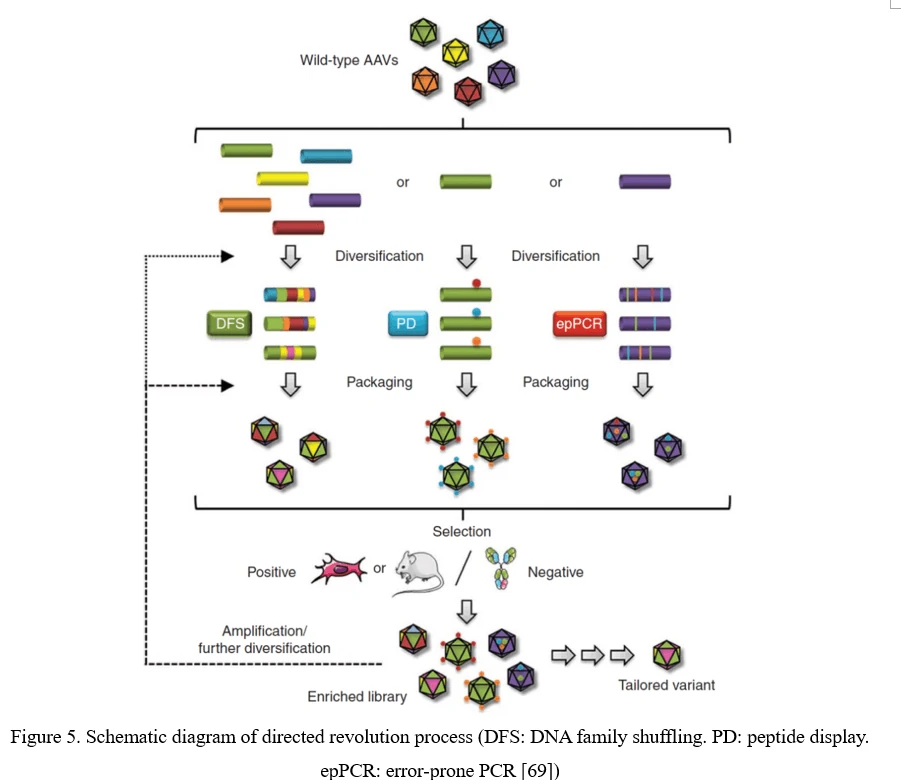
Point Mutation (Error-Prone PCR)
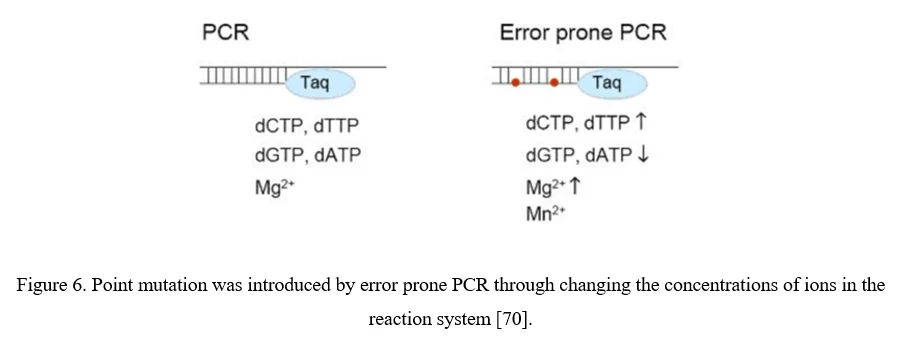
The simplest strategy for AAV diversification is mutagenesis of individual residues in the AAV viral capsid, either in a site-directed or in a randomized manner, expecting the introduced amino acid changes may alter essential AAV vector properties. Randomized mutations can be introduced to AAV capsid-encoding sequence by error-prone PCR (Figure 6).
Although point mutations are easily introduced to AAV capsid-encoding sequence, there still exist problems. 1) Only 0.01–1% of AAV mutations are typically beneficial; 2) By far, number of clones to cover all combinations of multiple mutations within a single AAV capsid exceeds technical possibilities. It would be more useful to focus on a specific region of the AAV capsid, especially the surface-exposed areas and AAV capsid protrusions which are more likely to interact with cellular receptors and antibodies [70].
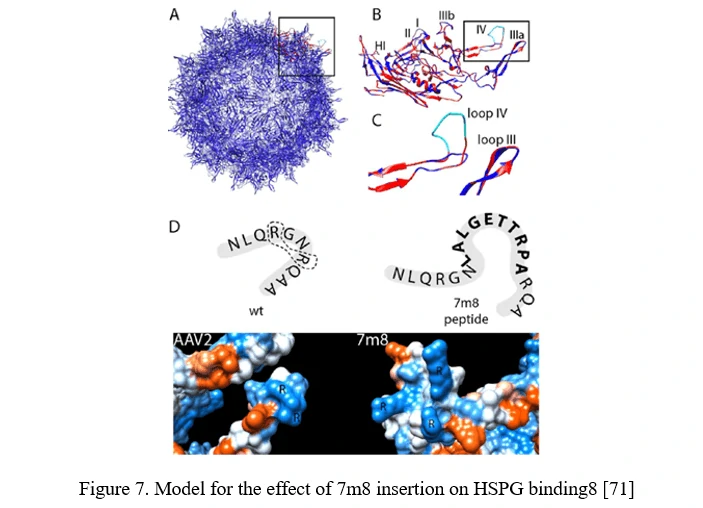
Peptide Display
Peptide insertion of novel receptor-binding elements on the AAV capsid surface can provide affinity for the receptor specifically expressed in the target cell type. To maximize success of this AAV peptide display strategy, it is critical to capitalize on sites in the AAV capsid that mediate surface exposure of the peptide.
In the case of AAV2, it can be achieved by insertion before or after arginine 588 (R588), one of the two vital residues required for AAV2 binding to HSPG. In the AAV2-7m8 variant, 7m8 disrupts the HSPG-binding motif, taking the arginines apart (Figure 7) [71].
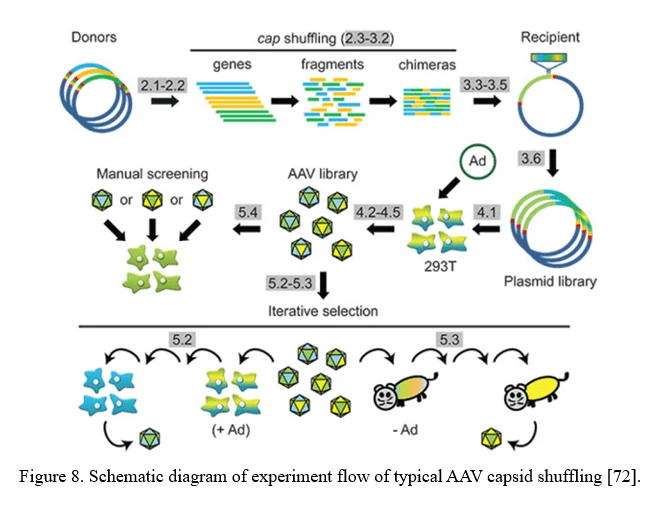
AAV family shuffling (DNA Shuffling of AAV capsids)
A chimeric capsid–encoding library can be generated by AAV family shuffling (Figure 8). Fragments of various AAV capsid genes are re-assembled based on partial homologies (typically >80% for most AAV serotypes). This library contains chimeras with broad diversity in cell tropism and neutralizing antibody resistance.
This approach can also harness and blend functions from newly identified AAV serotypes, or even rationally designed AAV capsid mutants, to potentially create a combinatorially limitless number of novel variants with new properties [72].
9. AAV Vector Transduction-AAV vector Gene Delivery in vivo
For normal tissues or organs, such as heart, liver, kidney, breast, pancreas, ovary, brain, eye, skeleton muscle, adipose tissue, etc., Genemedi systematically organizes the corresponding optimal AAV serotype, gene delivery method and injection volume for mouse and rat tissue infection. Recommended AAV gene delivery methods for different organs is shown in Table5.
| Infection organ | Recommended serotype | Injection route | Animal | Injection volume (μl) |
| Heart | AAV9 | Multiple points in situ | Rat | 10-15/point, 3-5 points |
| Mouse | 10-15/point, 3-5 points | |||
| Tail vein | Rat | 250 (200g body weight) | ||
| Mouse | 100 | |||
| Liver | AAV8 or AAV9 | Tail vein | Rat | 200 (200g body weight) |
| Mouse | 100 | |||
| Whole brain | AAV-PHP.eB AAV-PHP.B | Tail vein | Rat | 200 (200g body weight) |
| Mouse | 100 | |||
| Lateral ventricle | AAV9 | Stereotactic | Rat | 1-5 |
| Mouse | 1-5 | |||
| Brain tissue | AAV9 | Stereotactic | Rat | 2-3 |
| Mouse | 1-2 | |||
| Fat | AAV9 | Intraperitoneal injection for intraperitoneal fat | Rat | 300 |
| Mouse | 150-200 | |||
| in situ injection for subcutaneous fat | Rat | 10-15/point, 3-5 points | ||
| Mouse | 10-15/point, 3-5 points | |||
| Skeletal muscle | AAV1 or AAV9 | In situ injection | Rat | 10-15/point, 3-5 points |
| Mouse | 10-15/point, 3-5 points | |||
| Eyes | AAV2, AAV10 or AAV-DJ | vitreous chamber injection | Rat | 3-5 |
| Mouse | 1-3 | |||
| subretinal space injection | Rat | 1-2 | ||
| Mouse | 1-2 | |||
| Lung | AAV6 | Intratracheal injection | Rat | 100 (200g weight) |
| Mouse | 50-75 | |||
| Kidney | AAV2 or AAV9 | Renal pelvis injection | Rat | 10-15/point, 3-5 points |
| Mouse | 10-15/point, 3-5 points | |||
| Intestine | AAV1 or AAV5 | Enema | Rat | 200 (200g weight) |
| Mouse | 100 |
Detailed information of AAV in vivo animal injection protocol can be seen in AAV User Manual.
10. Adeno-associated virus (AAV vector) - Production Protocol, Guidelines And References
AAV Production Protocol–Genemedi
1. AAV plasmid construction
The gene of interest is cloned into one of the ITR/MCS-containing AAV vectors to generate AAV-GOI. The purity and RNAse contaminants of AAV viral plasmid should be taken into consideration.
2. AAV packaging
The recombinant AAV viral plasmid AAV-GOI is co-transfected into the AAV-293 cells with AAV helper plasmid pHelper (carrying adenovirus-derived genes) and AAV-RC (carrying AAV-2 replication and capsid genes), which together supply all the trans-acting factors required for AAV replication and packaging in the AAV-293 cells.
3. Harvesting AAV particles
Collecting the cells and freeze/thaw the cell pellet to release the AAV virus.
4. AAV purification
Purify the AAV virus with gradient ultracentrifugation to separate contaminants from the impure AAV preparations.
5.AAV titer detection
Determine AAV titer by real-time quantitative PCR (QPCR) using primers targeting the AAV ITR. Titer values are then identified by comparison to a standard curve of a plasmid sample of known concentration.
6.Quality control of AAV
After AAV virus titer detection, the infection activity needs to be evaluated before animal experiments by infecting cells such as 293T, CHO to test the gene expression. MOI for AAV mediated gene transduction in cells will be controlled ranging from 10^4 to 10^5.
Reference
1.Atchison RW, BC Casto and WM Hammon. (1965). Adenovirus-Associated Defective Virus Particles. Science 149:754-6.
2.Hoggan MD, NR Blacklow and WP Rowe. (1966). Studies of small DNA viruses found in various adenovirus preparations: physical, biological, and immunological characteristics. Proc Natl Acad Sci U S A 55:1467-74.
3.Wu Z, A Asokan and RJ Samulski. (2006). Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol Ther 14:316-27.
4.Vectors used in gene therapy clinical trials. The Journal of Gene Medicine Online Library. [Online] Updated Nov 2017. http://www.abedia.com/wiley/vectors.php.
5.Zsigmond E, K Kobayashi, KW Tzung, L Li, Y Fuke and L Chan. (1997). Adenovirus-mediated gene transfer of human lipoprotein lipase ameliorates the hyperlipidemias associated with apolipoprotein E and LDL receptor deficiencies in mice. Hum Gene Ther 8:1921-33.
6.Passini MA, J Bu, AM Richards, CM Treleaven, JA Sullivan, CR O’Riordan, A Scaria, AP Kells, L Samaranch, W San Sebastian, T Federici, MS Fiandaca, NM Boulis, KS Bankiewicz, LS Shihabuddin and SH Cheng. (2014). Translational fidelity of intrathecal delivery of self-complementary AAV9-survival motor neuron 1 for spinal muscular atrophy. Hum Gene Ther 25:619-30.
7.Stieger K and B Lorenz. (2014). [Specific gene therapy for hereditary retinal dystrophies – an update]. Klin Monbl Augenheilkd 231:210-5.
8.Trapani I, P Colella, A Sommella, C Iodice, G Cesi, S de Simone, E Marrocco, S Rossi, M Giunti, A Palfi, GJ Farrar, R Polishchuk and A Auricchio. (2014). Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol Med 6:194-211.
9.Doi K and Y Takeuchi. (2015). Gene therapy using retrovirus vectors: vector development and biosafety at clinical trials. Uirusu 65:27-36.
10.Duncan GA, N Kim, Y Colon-Cortes, J Rodriguez, M Mazur, SE Birket, SM Rowe, NE West, A Livraghi-Butrico, RC Boucher, J Hanes, G Aslanidi and JS Suk. (2018). An Adeno-Associated Viral Vector Capable of Penetrating the Mucus Barrier to Inhaled Gene Therapy. Mol Ther Methods Clin Dev 9:296-304.
11.Bowles DE, SW McPhee, C Li, SJ Gray, JJ Samulski, AS Camp, J Li, B Wang, PE Monahan, JE Rabinowitz, JC Grieger, L Govindasamy, M Agbandje-McKenna, X Xiao and RJ Samulski. (2012). Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol Ther 20:443-55.
12.Nathwani AC, EG Tuddenham, S Rangarajan, C Rosales, J McIntosh, DC Linch, P Chowdary, A Riddell, AJ Pie, C Harrington, J O’Beirne, K Smith, J Pasi, B Glader, P Rustagi, CY Ng, MA Kay, J Zhou, Y Spence, CL Morton, J Allay, J Coleman, S Sleep, JM Cunningham, D Srivastava, E Basner-Tschakarjan, F Mingozzi, KA High, JT Gray, UM Reiss, AW Nienhuis and AM Davidoff. (2011). Adeno-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 365:2357-65.
13.Jessup M, B Greenberg, D Mancini, T Cappola, DF Pauly, B Jaski, A Yaroshinsky, KM Zsebo, H Dittrich, RJ Hajjar and I Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease. (2011). Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 124:304-13.
14.LeWitt PA, AR Rezai, MA Leehey, SG Ojemann, AW Flaherty, EN Eskandar, SK Kostyk, K Thomas, A Sarkar, MS Siddiqui, SB Tatter, JM Schwalb, KL Poston, JM Henderson, RM Kurlan, IH Richard, L Van Meter, CV Sapan, MJ During, MG Kaplitt and A Feigin. (2011). AAV2-GAD gene therapy for advanced Parkinson’s disease: a double-blind, sham-surgery controlled, randomised trial. Lancet Neurol 10:309-19.
15.Mease PJ, N Wei, EJ Fudman, AJ Kivitz, J Schechtman, RG Trapp, KF Hobbs, M Greenwald, A Hou, SA Bookbinder, GE Graham, CW Wiesenhutter, L Willis, EM Ruderman, JZ Forstot, MJ Maricic, KH Dao, CH Pritchard, DN Fiske, FX Burch, HM Prupas, P Anklesaria and AE Heald. (2010). Safety, tolerability, and clinical outcomes after intraarticular injection of a recombinant adeno-associated vector containing a tumor necrosis factor antagonist gene: results of a phase 1/2 Study. J Rheumatol 37:692-703.
16.Evans CH, SC Ghivizzani and PD Robbins. (2018). Gene Delivery to Joints by Intra-Articular Injection. Hum Gene Ther 29:2-14.
17.Gupta PR and RM Huckfeldt. (2017). Gene therapy for inherited retinal degenerations: initial successes and future challenges. J Neural Eng 14:051002.
18.Polinski NK, SE Gombash, FP Manfredsson, JW Lipton, CJ Kemp, A Cole-Strauss, NM Kanaan, K Steece-Collier, NC Kuhn, SL Wohlgenant and CE Sortwell. (2015). Recombinant adenoassociated virus 2/5-mediated gene transfer is reduced in the aged rat midbrain. Neurobiol Aging 36:1110-20.
19.Moore T. (2005). Restoring retinal function in a mouse model of hereditary blindness. PLoS Med 2:e399.
20.Russell S, J Bennett, JA Wellman, DC Chung, ZF Yu, A Tillman, J Wittes, J Pappas, O Elci, S McCague, D Cross, KA Marshall, J Walshire, TL Kehoe, H Reichert, M Davis, L Raffini, LA George, FP Hudson, L Dingfield, X Zhu, JA Haller, EH Sohn, VB Mahajan, W Pfeifer, M Weckmann, C Johnson, D Gewaily, A Drack, E Stone, K Wachtel, F Simonelli, BP Leroy, JF Wright, KA High and AM Maguire. (2017). Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 390:849-860.
21.Dulsat C. (2017). American Academy of Neurology – 69th Annual Meeting (April 22-28, 2017 – Boston, Massachusetts, USA). Drugs Today (Barc) 53:309-319.
22.Annexstad EJ, I Lund-Petersen and M Rasmussen. (2014). Duchenne muscular dystrophy. Tidsskr Nor Laegeforen 134:1361-4.
23.Blankinship MJ, P Gregorevic and JS Chamberlain. (2006). Gene therapy strategies for Duchenne muscular dystrophy utilizing recombinant adeno-associated virus vectors. Mol Ther 13:241-9.
24.Kawecka K, M Theodoulides, Y Hasoglu, S Jarmin, H Kymalainen, A Le-Heron, L Popplewell, A Malerba, G Dickson and T Athanasopoulos. (2015). Adeno-Associated Virus (AAV) Mediated Dystrophin Gene Transfer Studies and Exon Skipping Strategies for Duchenne Muscular Dystrophy (DMD). Curr Gene Ther 15:395-415.
25.Kodippili K, CH Hakim, X Pan, HT Yang, Y Yue, Y Zhang, JH Shin, NN Yang and D Duan. (2018). Dual AAV Gene Therapy for Duchenne Muscular Dystrophy with a 7-kb Mini-Dystrophin Gene in the Canine Model. Hum Gene Ther 29:299-311.
26.Duan D. (2018). Systemic AAV Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol Ther 26:2337-2356.
27.Santagostino E and MR Fasulo. (2013). Hemophilia a and hemophilia B: different types of diseases? Semin Thromb Hemost 39:697-701.
28.Nichols T, MH Whitford, VR Arruda, HH Stedman, MA Kay and KA High. (2014). Translational Data from AAV-Mediated Gene Therapy of Hemophilia B in Dogs. Hum Gene Ther Clin Dev.
29.Crudele JM, JD Finn, JI Siner, NB Martin, GP Niemeyer, S Zhou, F Mingozzi, CD Lothrop, Jr. and VR Arruda. (2015). AAV liver expression of FIX-Padua prevents and eradicates FIX inhibitor without increasing thrombogenicity in hemophilia B dogs and mice. Blood 125:1553-61.
30.Hasbrouck NC and KA High. (2008). AAV-mediated gene transfer for the treatment of hemophilia B: problems and prospects. Gene Ther 15:870-5.
31.Brimble MA, UM Reiss, AC Nathwani and AM Davidoff. (2016). New and improved AAVenues: current status of hemophilia B gene therapy. Expert Opin Biol Ther 16:79-92.
32.Grieger JC and RJ Samulski. (2005). Packaging capacity of adeno-associated virus serotypes: impact of larger genomes on infectivity and postentry steps. J Virol 79:9933-44.
33.Selot RS, S Hareendran and GR Jayandharan. (2014). Developing immunologically inert adeno-associated virus (AAV) vectors for gene therapy: possibilities and limitations. Curr Pharm Biotechnol 14:1072-82.
34.Bartlett JS, R Wilcher and RJ Samulski. (2000). Infectious entry pathway of adeno-associated virus and adeno-associated virus vectors. J Virol 74:2777-85.
35.Ding W, L Zhang, Z Yan and JF Engelhardt. (2005). Intracellular trafficking of adeno-associated viral vectors. Gene Ther 12:873-80.
36.Srivastava A. (2008). Adeno-associated virus-mediated gene transfer. J Cell Biochem 105:17-24.
37.Berns KI. (1990). Parvovirus replication. Microbiol Rev 54:316-29.
38.Pereira DJ, DM McCarty and N Muzyczka. (1997). The adeno-associated virus (AAV) Rep protein acts as both a repressor and an activator to regulate AAV transcription during a productive infection. J Virol 71:1079-88.
39.Calcedo R and JM Wilson. (2013). Humoral Immune Response to AAV. Front Immunol 4:341.
40.Mingozzi F and KA High. (2013). Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood 122:23-36.
41.Weitzman MD and RM Linden. (2011). Adeno-associated virus biology. Methods Mol Biol 807:1-23.
42.Gao G, MR Alvira, S Somanathan, Y Lu, LH Vandenberghe, JJ Rux, R Calcedo, J Sanmiguel, Z Abbas and JM Wilson. (2003). Adeno-associated viruses undergo substantial evolution in primates during natural infections. Proc Natl Acad Sci U S A 100:6081-6.
43.Hauck B, L Chen and W Xiao. (2003). Generation and characterization of chimeric recombinant AAV vectors. Mol Ther 7:419-25.
44.Rabinowitz JE, DE Bowles, SM Faust, JG Ledford, SE Cunningham and RJ Samulski. (2004). Cross-dressing the virion: the transcapsidation of adeno-associated virus serotypes functionally defines subgroups. J Virol 78:4421-32.
45.Manno CS, AJ Chew, S Hutchison, PJ Larson, RW Herzog, VR Arruda, SJ Tai, MV Ragni, A Thompson, M Ozelo, LB Couto, DG Leonard, FA Johnson, A McClelland, C Scallan, E Skarsgard, AW Flake, MA Kay, KA High and B Glader. (2003). AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood 101:2963-72.
46.Bartlett JS, RJ Samulski and TJ McCown. (1998). Selective and rapid uptake of adeno-associated virus type 2 in brain. Hum Gene Ther 9:1181-6.
47.Richter M, A Iwata, J Nyhuis, Y Nitta, AD Miller, CL Halbert and MD Allen. (2000). Adeno-associated virus vector transduction of vascular smooth muscle cells in vivo. Physiol Genomics 2:117-27.
48.Koeberl DD, IE Alexander, CL Halbert, DW Russell and AD Miller. (1997). Persistent expression of human clotting factor IX from mouse liver after intravenous injection of adeno-associated virus vectors. Proc Natl Acad Sci U S A 94:1426-31.
49.Pajusola K, M Gruchala, H Joch, TF Luscher, S Yla-Herttuala and H Bueler. (2002). Cell-type-specific characteristics modulate the transduction efficiency of adeno-associated virus type 2 and restrain infection of endothelial cells. J Virol 76:11530-40.
50.Qing K, C Mah, J Hansen, S Zhou, V Dwarki and A Srivastava. (1999). Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus 2. Nat Med 5:71-7.
51.Summerford C and RJ Samulski. (1998). Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol 72:1438-45.
52.Summerford C, JS Bartlett and RJ Samulski. (1999). AlphaVbeta5 integrin: a co-receptor for adeno-associated virus type 2 infection. Nat Med 5:78-82.
53.Shi W, A Hemminki and JS Bartlett. (2006). Capsid modifications overcome low heterogeneous expression of heparan sulfate proteoglycan that limits AAV2-mediated gene transfer and therapeutic efficacy in human ovarian carcinoma. Gynecol Oncol 103:1054-62.
54.Wang Z, T Zhu, C Qiao, L Zhou, B Wang, J Zhang, C Chen, J Li and X Xiao. (2005). Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol 23:321-8.
55.Nakai H, S Fuess, TA Storm, S Muramatsu, Y Nara and MA Kay. (2005). Unrestricted hepatocyte transduction with adeno-associated virus serotype 8 vectors in mice. J Virol 79:214-24.
56.Gao G, LH Vandenberghe, MR Alvira, Y Lu, R Calcedo, X Zhou and JM Wilson. (2004). Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol 78:6381-8.
57.Saraiva J, RJ Nobre and L Pereira de Almeida. (2016). Gene therapy for the CNS using AAVs: The impact of systemic delivery by AAV9. J Control Release 241:94-109.
58.Alisky JM, SM Hughes, SL Sauter, D Jolly, TW Dubensky, Jr., PD Staber, JA Chiorini and BL Davidson. (2000). Transduction of murine cerebellar neurons with recombinant FIV and AAV5 vectors. Neuroreport 11:2669-73.
59.Burger C, OS Gorbatyuk, MJ Velardo, CS Peden, P Williams, S Zolotukhin, PJ Reier, RJ Mandel and N Muzyczka. (2004). Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol Ther 10:302-17.
60.Davidson BL, CS Stein, JA Heth, I Martins, RM Kotin, TA Derksen, J Zabner, A Ghodsi and JA Chiorini. (2000). Recombinant adeno-associated virus type 2, 4, and 5 vectors: transduction of variant cell types and regions in the mammalian central nervous system. Proc Natl Acad Sci U S A 97:3428-32.
61.Gao GP, MR Alvira, L Wang, R Calcedo, J Johnston and JM Wilson. (2002). Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci U S A 99:11854-9.
62.Chao H, Y Liu, J Rabinowitz, C Li, RJ Samulski and CE Walsh. (2000). Several log increase in therapeutic transgene delivery by distinct adeno-associated viral serotype vectors. Mol Ther 2:619-23.
63.Blankinship MJ, P Gregorevic, JM Allen, SQ Harper, H Harper, CL Halbert, AD Miller and JS Chamberlain. (2004). Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol Ther 10:671-8.
64.Halbert CL, JM Allen and AD Miller. (2001). Adeno-associated virus type 6 (AAV6) vectors mediate efficient transduction of airway epithelial cells in mouse lungs compared to that of AAV2 vectors. J Virol 75:6615-24.
65.Kotterman MA and DV Schaffer. (2014). Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet 15:445-51.
66.Zhong L, B Li, CS Mah, L Govindasamy, M Agbandje-McKenna, M Cooper, RW Herzog, I Zolotukhin, KH Warrington, Jr., KA Weigel-Van Aken, JA Hobbs, S Zolotukhin, N Muzyczka and A Srivastava. (2008). Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc Natl Acad Sci U S A 105:7827-32.
67.Lochrie MA, GP Tatsuno, B Christie, JW McDonnell, S Zhou, R Surosky, GF Pierce and P Colosi. (2006). Mutations on the external surfaces of adeno-associated virus type 2 capsids that affect transduction and neutralization. J Virol 80:821-34.
68.Munch RC, H Janicki, I Volker, A Rasbach, M Hallek, H Buning and CJ Buchholz. (2013). Displaying high-affinity ligands on adeno-associated viral vectors enables tumor cell-specific and safe gene transfer. Mol Ther 21:109-18.
69.Grimm D and S Zolotukhin. (2015). E Pluribus Unum: 50 Years of Research, Millions of Viruses, and One Goal–Tailored Acceleration of AAV Evolution. Mol Ther 23:1819-31.
70.Maheshri N, JT Koerber, BK Kaspar and DV Schaffer. (2006). Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat Biotechnol 24:198-204.
71.Khabou H, M Desrosiers, C Winckler, S Fouquet, G Auregan, AP Bemelmans, JA Sahel and D Dalkara. (2016). Insight into the mechanisms of enhanced retinal transduction by the engineered AAV2 capsid variant -7m8. Biotechnol Bioeng 113:2712-2724.
72.Kienle E, E Senis, K Borner, D Niopek, E Wiedtke, S Grosse and D Grimm. (2012). Engineering and evolution of synthetic adeno-associated virus (AAV) gene therapy vectors via DNA family shuffling. J Vis Exp.
