





Gene editing, also called genome editing, is a group of technologies to change the sequence of DNA in the genome. Several approaches to genome editing have been developed, including Zinc Finger, TALEN, and CRISPR/Cas9. Compared with the other gene editing tools, CRISPR/Cas9 system is faster, cheaper, more accurate, and more efficient, showing unprecedented advantages in gene therapy.
CRISPR (clustered regularly interspaced short palindromic repeats) is a family of DNA sequences derived from viruses that have previously infected the prokaryotes and has the ability to recognize invasive homologous DNA sequences from similar viruses, then directing Cas9 (CRISPR-associated 9) nuclease to specifically cleave them during subsequent infections, thus playing a key role in the antiviral defense system of prokaryotes [4]. Cas9 nuclease is an enzyme guided by CRISPR sequences to recognize and cleave specific nucleic acids which are complementary to the CRISPR sequence [5]. CRISPR sequences together with Cas9 enzymes make up the basis of a CRISPR/Cas9 technology which is a versatile genome-editing platform within organisms.
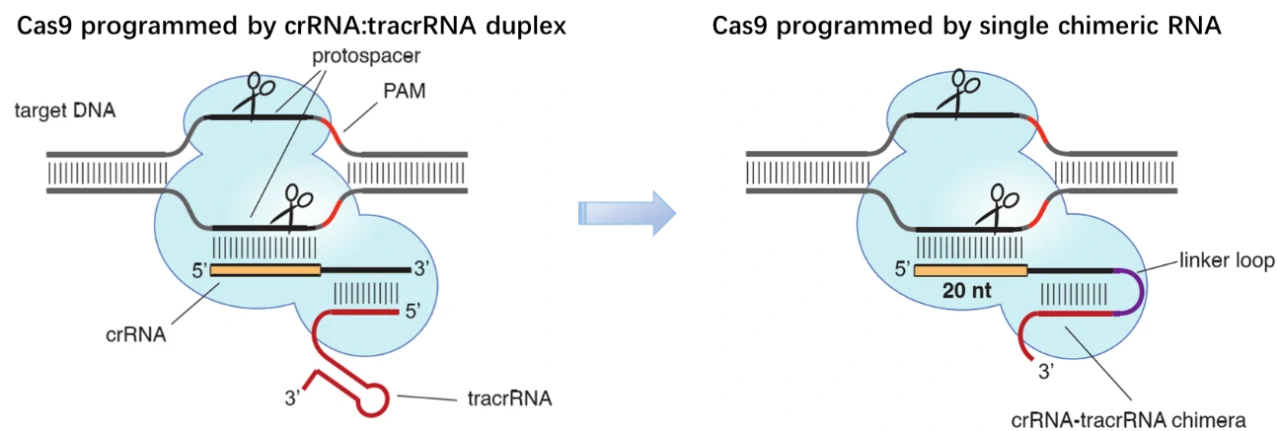
By 2010, three CRISPR/Cas9 systems have been identified in bacteria: type I, II and III. Derived from Streptococcus pyogenes, type II CRISPR system is relatively simple and has been eventually adapted for genome editing in mammalian cells [1]. This simpler CRISPR/Cas9 system contains four components, i.e. Cas9 endonuclease, trans-activating CRISPR RNA (tracrRNA) and two small RNA molecules named CRISPR RNA (crRNA) [2]. In this system, the mature crRNA base-paired to trans-activating crRNA (tracrRNA) forms a two-RNA structure, guiding Cas9 nuclease to mediate the recognition and cleavage of target nucleic acids [3-5]. At sites complementary to the crRNA-guide sequence, the HNH nuclease domain of Cas9 cleaves the complementary strand, whereas the RuvC-like domain of Cas9 cleaves the non-complementary strand. To target nucleic acids with more precision and expand the application of CRISPR/Cas9 system, great efforts have been paid to engineer the Cas9 enzyme or alter PAM specificities. To date, a wide range of Cas9 derivatives with novel PAM specificities have been generated using the site-depletion assay [6], and the initial two-RNA structure, ie. CRISPR RNAs (crRNAs) base-paired to trans-activating crRNA (tracrRNA), have been programmed into a “single-guide RNA (sgRNA)” (dual-tracrRNA:crRNA) by fusing the 3′ end of crRNA to the 5′ end of tracrRNA [7]. By manipulating the nucleotide sequence of the sgRNA, the artificial CRISPR/Cas9 system could be programmed to target any DNA sequence for cleavage [7].
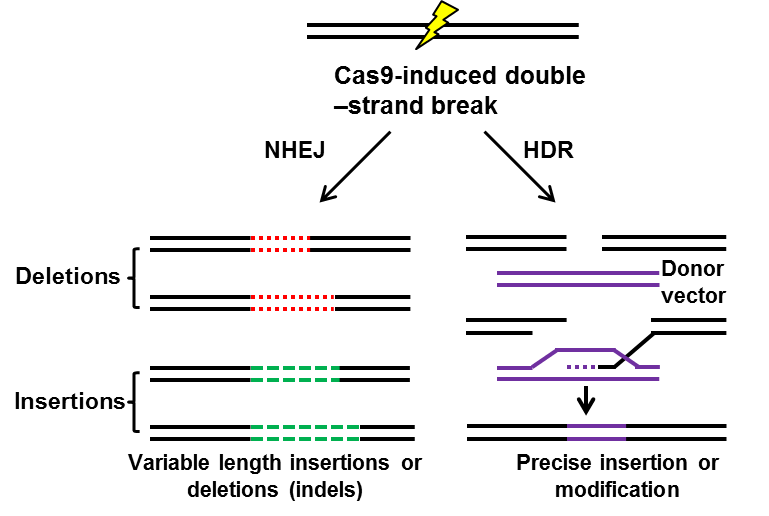
Guided by small guide RNA (sgRNA), Cas9 nuclease cleaves double-strand DNA, the inducing site-specific genomic double-strand breaks (DSBs). In mammalian cells, there are 2 manners with which DSBs are repaired: non-homologous end joining (NHEJ) or homology directed repair (HDR). (I) Frequently, NHEJ happens, enabling the introduction of small deletions or insertions between the ends of DSBs and resulting in frameshift mutations, and thus mediates gene knockout [8,9]. (II) With the existence of repair template, such as exogenous donor vectors, HDR takes place, introducing precise gene modifications, such as reporter insertions, and thus mediate gene knockin [10,11]. Both NHEJ mediated gene knockout HDR mediated gene knockin have been widely used in the field of gene therapy [12]. However, HDR (0.5%-20%) is much less frequent than NHEJ (20%-60%) [13], and the former only occurs during S and G2 phase while the latter can occur at any time of cell cycle [14-16]. To improve the efficiency of HDR, people have screened so many small molecules, finding that small molecules inhibitor Scr7, targeting DNA ligase IV, one key enzyme in the NHEJ pathway, significantly facilitates the HDR pathway (5%-58.3%) [17]. So far, NHEJ is widely used in the mediation of gene knockout [8,18] and correction of pathogenic gene mutation [12,19,20], while HDR is applied for knockin schemes, such as knockin cell line generation [21] or reporter/tag knockin [22].
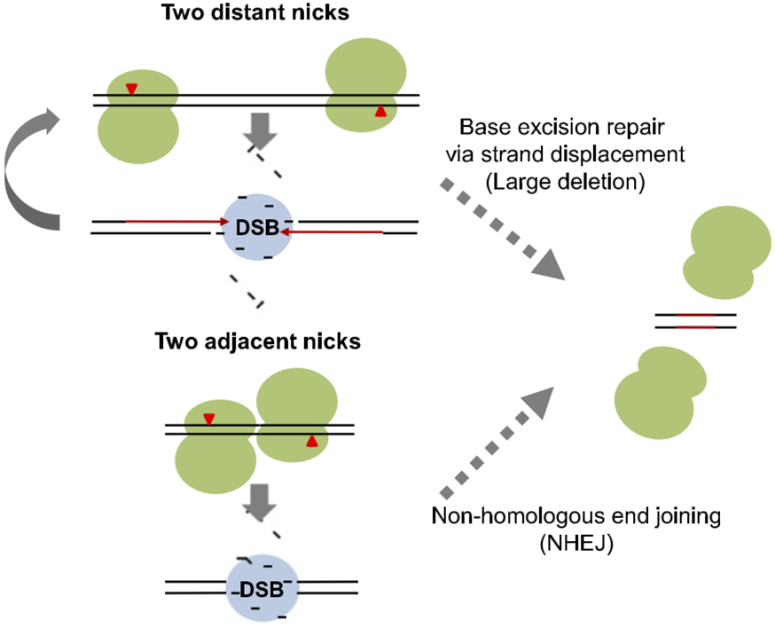
Although CRISPR/Cas9 system is much cheaper, more efficient and flexible, high efficiency of off-target mutations or unwanted DNA cleavages have limited the application of this system. Mutation exploration revealed that Cas9 nickase carrying the mutation of D10A (D10A Cas9) loses the nuclease activity in RuvC domain, but does not affect the nuclease activity of HNH domain. In combination with paired gRNAs, D10A Cas9 cuts target DNA and generates two single-strand breaks with higher specificity and little off-target mutations, which is confirmed by exome sequencing analysis [25,26]. By comparing gene-disrupting efficiency of Cas9 paired nickases with that of nuclease, the T7E1 assay and deep sequencing revealed comparable on-target efficiency, even sometimes higher on-target efficiency [27]. Moreover, FACS enrichment of paired D10A Cas9 nickases will significantly facilitate efficient gene editing with minimized off-target effects [28]. Further modifications showed that hRad51–Cas9 nickase, generated by hRad51 mutants fusion with Cas9 (D10A) nickase (RDN), mediates HDR without double-stranded breaks, which results in higher HDR:indel ratios and lower off-target activity than Cas9 nuclease [29].
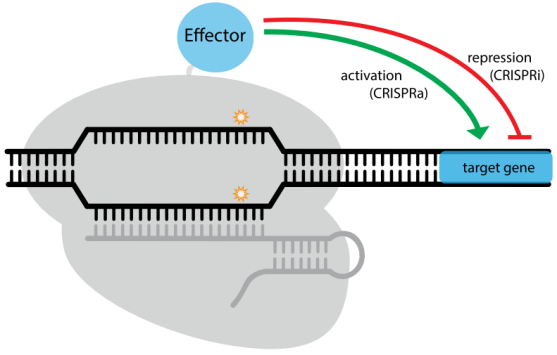
The nuclease-deactivated Cas9 variant, dCas9, was generated by introducing two mutations (D10A and H840A) (Figure. 2A), resulting in no catalytic activity in both RuvC domain and HNH domain [30]. With great DNA binding activity, dCas9 has been widely applied to regulate gene expression in combination with transcription activator or repressor [14,31] (Table 3). (I) dCas9 activation system: transcription activator domains (VP64, p65, MPH) or epigenetic activation modifications (p300, CBP etc.), have been fused to dCas9 to activate gene expression [32-35]; (II) dCas9 repression system: transcription repressor domains (KRAB) or epigenetic repression modifications (LSD1, DNMT3A etc.), have been fused to down-regulate the expression of target genes [34,36-38]. Besides the direct effector fusion manner, functional scaffolds (such as SunTag) can be incorporated into dCas9/sgRNA complex to recruit effectors to affect transcription [36,39]. Spatiotemporal control in dCas9/sgRNA-induced effector recruitment was also achieved upon chemical or light induction [40,41].
| Table 1. Effectors in dCas9 systems | ||
| Effector | Transcription regulation | |
| Activator | VP64 | transcription activation |
| Activator | VP48 | transcription activation |
| Activator | VP120 | transcription activation |
| Activator | p65 | transcription activation |
| Activator | MPH (MS2-P65-HSF1) | transcription activation |
| Activator | P65-HSF1 | transcription activation |
| Activator | VP64-p65-Rta | transcription activation |
| Repressor | KRAB | transcription repression |
| Repressor | SID4x | transcription repression |
| Repressor | Mxi1 | transcription repression |
| Histone acetylation | p300 | transcription activation |
| Histone acetylation | CBP | transcription activation |
| Histone demethylation | LSD1 | transcription repression |
| DNA methylation | DNMT3A | transcription repression |
| DNA demethylation | TET1 | transcription activation |
The traditional CRISPR/Cas9 system requires specific PAM sequence, which might be an obstacle to perform gene editing in some cases. Moreover, the off-target effect and cytotoxicity also limit the application of CRISPR/Cas9 system. To overcome these difficulties, researchers successfully screened several other tools to perfect the gene editing system, such as CRISPR/Cas13 and CRISPR/Cpf1. etc.
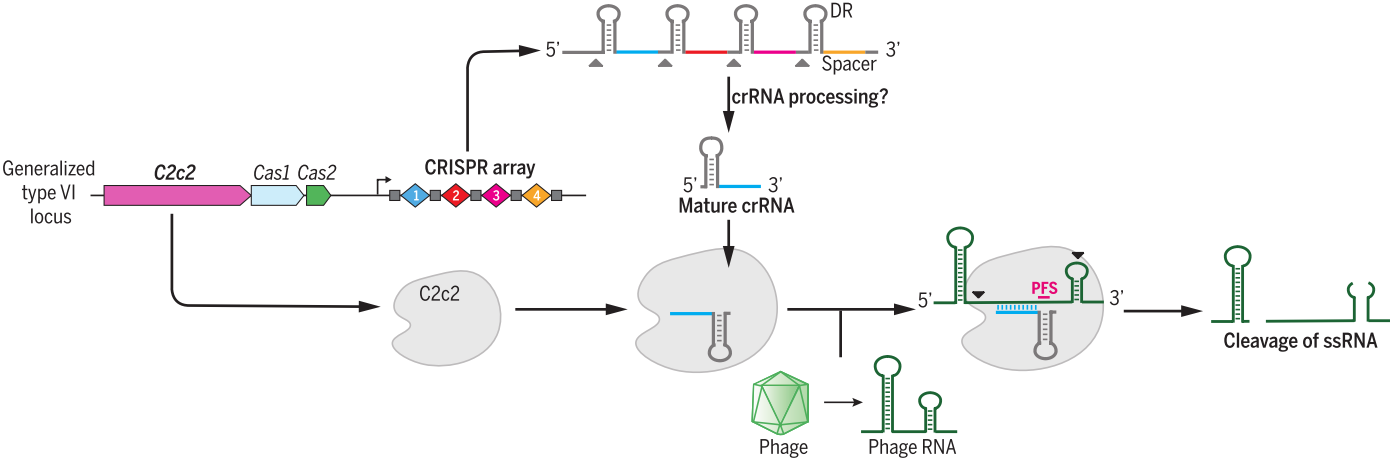
Cas13 family, containing Cas13a (also known as C2c2), Cas13b and Cas13c [43], belongs to class 2 type VI and have two higher eukaryotes and prokaryotes nucleotide-binding (HEPN) endoRNase domains to mediate precise RNA cleavage in targets with protospacer flanking sites [44,45]. Cas13 enzymes are programmable and have been applied for nucleic acid detection as well as RNA knockdown and transcript tracking [46,47]. Combined with base editor, such as ADAR2 (adenosine deaminase acting on RNA type 2), catalytically inactive Cas13 (dCas13) have been used as guidance with no strict sequence constraints to direct RNA base editing, which is a promising platform with great applicability for both basic research and clinical therapy [48].
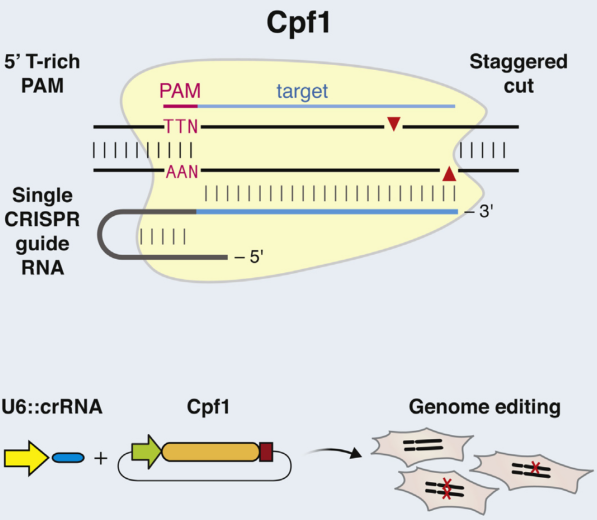
Distinct with Cas9 enzyme, Cpf1 is from class 2 type V CRISPR system (Prevotella and Francisella 1) and mediates DNA cleavage with three features. 1) The guiding sequence is processed into mature crRNA with no requirement of the tracrRNA. 2) Guided by mature crRNA, Cpf1 efficiently cut the DNA with a short T-rich protospacer-adjacent motif (PAM), not the G-rich PAM in Cas9 system. 3) Cpf1-crRNA system generates a staggered DNA double-stranded break with a 4 or 5-nt 5’ overhang [49]. These features greatly expand the application of CRISPR system [50].

