CRISPR/Cas9 system – gRNA design and validation – principle and protocol

DIAGNOSTIC ANTIBODY & ANTIGEN
SERVICES
- Custom AAV Production
- Custom Adenovirus Production
- Custom Lentivirus Production
- Pre-made AAV Production
- Pre-made Adenovirus Production
- Pre-made Lentivirus Production
- AAV-LC3 Autophagy Flux Detection
- Ad-LC3 Autophagy Flux Detection
- Lv-LC3 Autophagy Flux Detection
- AAV Control Virus Production
- CRISPR/Cas9 AAV Production
- CRISPR/Cas9 Adenovirus Production

Gene editing, also called genome editing, is a group of technologies to change the sequence of DNA in the genome. Several approaches to genome editing have been developed, including Zinc Finger, TALEN, and CRISPR/Cas9. Compared with the other gene editing tools, CRISPR/Cas9 system is faster, cheaper, more accurate, and more efficient, showing unprecedented advantages in gene therapy.
CRISPR/Cas9 premade products
Cat No. | Products Name | Type of Crispr | Viral vector | Promoter | Fluorescent/Resistance | Tag | Order
|
|---|---|---|---|---|---|---|---|
spCas9 |
lentivirus |
CBH |
EGFP |
FLAG |
|||
spCas9 |
lentivirus |
CBH |
mcherry |
FLAG |
|||
AAV-CMV-saCas9 |
saCas9 |
AAV |
CMV |
Null |
HA |

Cas9-gRNA design principles:
1) The PAM sequence that SpCas9 recognizes is NGG (AGG, TGG, CGG, GGG), while SaCas9 recognizes NNGRRT (NNGAAT, NNGAGT, NNGGAT, NNGGGT) or NNGRRN. For sgRNA, the length is about 21 or 22 nucleotides.
2) For sgRNA, the GC content in 40%~60% is better.
3) If the sgRNA is driven by U6 or T7 promoter, the 5’ end of sgRNA can be designed as G or GG to improve transcription efficiency, which should be considered.
4) The binding site of sgRNA should be as close to the coding region in the downstream of ATG as possible to induce frameshift mutation, the first or second exon is better.
5) SNPs should be checked in the binding site of sgRNA.
6) The distance between paired sgRNA should be taken into consideration before designing paired-gRNAs, if using Cas9 single nickase.
7) Whole genome off-target effect analysis is suggested. At least 5 bases can be allowed for the base mismatch and whether the off target is located in the gene encoding regions need to be confirmed. What’s more, base insertion or deletion in off targets should be detected.
To date, there exist so many CRISPR/Cas9 design tools, and some common tools are list in table 1.
| Name | Web | Affiliation |
| CRISPR design | http://crispr.mit.edu/ | Massachusetts Institute of Technology |
| E-Crisp | www.e-crisp.org/E-CRISP/designcrispr | DKFZ German Cancer Research Center |
| CRISPR design tool | http://www.broadinstitute.org/mpg/crispr_design/ | The Broad Institute of Harvard and MIT |
| Cas-OFFinder | http://www.rgenome.net | Harvard Medical School |
| CROP-IT | http://www.adlilab.org/CROP-IT/homepage.html | University of Virginia |
| GPP sgRNA Designer | https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design | The Broad Institute of Harvard and MIT |
1)SpCas9 gRNA design, database and validation
Based on the above principles, SpCas9-gRNA can be designed on the following website [64]: https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design. Choose the CRISPR enzyme type “SpyoCas9 (NGG)”, then input transcript IDs, Gene IDs/Symbols, raw DNA sequence or upload a list of transcript IDs, Gene IDs/Symbols, a FASTA file of DNA sequences. Click on the “submit” button, and you can get and download the gRNA sequence from the following website.
Synthesize the SpCas9 gRNA sequence and its anti-sense sequence, anneal the sequences into a double chain and clone into pLv-SpCas9 vector or pAd-SpCas9 vector, which is confirmed by Sanger sequencing.
Deliver the pLv-SpCas9 vector or pAd-SpCas9 vector containing target sgRNA sequences into cells, and verify knockout efficiency.
2)SaCas9 gRNA design, database and validation
Based on the above principles, SaCas9-gRNA can be designed on the following website [64]: https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design. Choose the CRISPR enzyme type “SaurCas9 (NNGRR)”, then input transcript IDs, Gene IDs/Symbols, raw DNA sequence or upload a list of transcript IDs, Gene IDs/Symbols, a FASTA file of DNA sequences. Click on the “submit” button, and you can get and download the gRNA sequence from the following website.
Synthesize the SaCas9 gRNA sequence and its anti-sense sequence, anneal the sequences into a double chain and clone into pAAV-SaCas9 vector, which is confirmed by Sanger sequencing.
Deliver the pAAV-SaCas9 vector containing target sgRNA sequences into cells, and verify knockout efficiency.
3)Summary of approaches for gRNA validation
The cutting efficiency of Cas9 nuclease is mainly affected gRNA target efficiency, which can be validated by T7 endonuclease I (T7EI) assay, Sanger sequencing and Western blot.
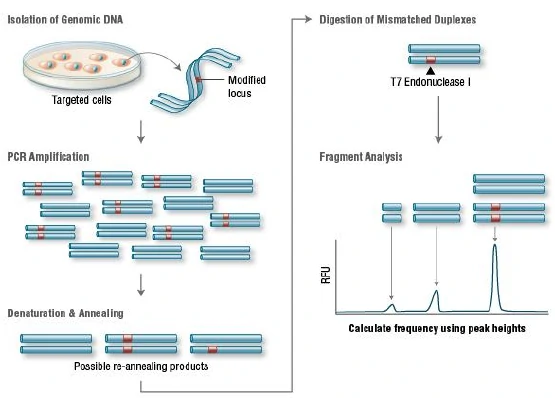
a.T7 endonuclease I (T7EI) assay
T7 Endonuclease I recognizes and cleaves non-perfectly matched DNA, which is a simple and cheap method to determine genome targeting efficiency of CRISPR/Cas9 system. First, extract the genomic DNA of cells whose genomes were targeted by Cas9. Second, perform PCR experiments to amplify target DNA. Third, anneal the PCR products and digest them with T7 Endonuclease I. Fourth, analyze the fragments with agarose gel to determine the efficiency of gRNA targeting.
b.Sanger sequencing assay
Sanger sequencing is a direct and precise way to examine the target efficiency of gRNA. First, extract the genomic DNA of cells whose genomes were targeted by Cas9. Second, perform PCR experiments to amplify target DNA. Third, verify the sequence of target genome by Sanger sequencing and then calculate the cleavage efficiency.
c.Western blot (WB) assay
Western blot is also simple, easy but no so precise assay for the determination of gRNA targeting efficiency. First, extract the whole cell protein of cells whose genomes were targeted by Cas9. Second, perform Western blot experiments to detect the expression levels of target protein. Third, confirm the efficiency of gRNA by analyzing the WB results.
4)How to detect the off-target?
Although CRISPR/Cas9 system is a versatile gene editing technology that is widely applied in the study of basic science and translational research, high frequency of off-target activity is really a restriction factor for its therapeutic and clinical applications. So far, several methods have been used to determine the off-targets with different advantages and disadvantages [65].
|
Table 5. Common CRISPR/Cas9 design tools [65] |
|||
| Method | Advantage | Disadvantage | References |
| T7E1 assay | Simple | Poor sensitivity, not cost-effective | [66,67] |
| Deep sequencing | Precise | Biased, misses potential off-target sites elsewhere in the genome | [25,68] |
| In silico prediction | Predicts some off-target mutation sites | Fails to predict bona-fide off-target sites | [69,70] |
| ChIP-seq | Unbiased detection of Cas9 binding sites genome-wide | Most off-target DNA-binding sites recognized by dCas9 are not cleaved at all by Cas9 in cells | [71-74] |
| GUIDE-seq | Unbiased, sensitive (0.1%), qualitative translocations, identifies breakpoint hotspots | False negatives present, limited by chromatin accessibility | [75] |
| HTGTS | Identifies translocations | False negatives present, limited by chromatin accessibility | [73] |
| IDLV | Programmable, sensitive (1%) | Many bona-fide off-target sites cannot be captured | [76] |
| Digenome-seq | Sensitive (0.1% or lower), unbiased and cost-effective | Not widely used | [77] |
| FISH | Quick | Less precise | [78] |
ChIP-seq, Chromatin immunoprecipitation followed by massively parallel DNA sequencing
GUIDE-seq, Genome-wide unbiased identification of DSBs enabled by sequencing
HTGTS, high-throughput, genome-wide, translocation sequencing
IDLV, integrase-defective lentiviral vectors
FISH, fluorescence in situ hybridization
5)How to minimize off-target?
a. Alter the sgRNA sequence. Truncation of the 3′ end of sgRNA, shortening the region complementary to the target site at the 5′ end of the sgRNA by as many as 3 nt (tru-gRNA) or addition of two guanine nucleotides to the 5′ end of the sgRNA improves target specificity [79].
b. Try to optimize the design of Cas9 and sgRNA design with design tools or directed evolution [64,80].
c. Purified Cas9 ribonucleoprotein works more immediately than plasmids containing Cas9 gene sequence and can be degraded rapidly in cells, which may help minimize the off-target frequency [81,82].
d. Strategies that restrict the transcription and translation of Cas9 may help minimize off-target effects [83].
e. Replace the wild-type Cas9 nuclease with D10 mutant Cas9 nickase and paired with two sgRNAs that each cleaves only one strand [28].
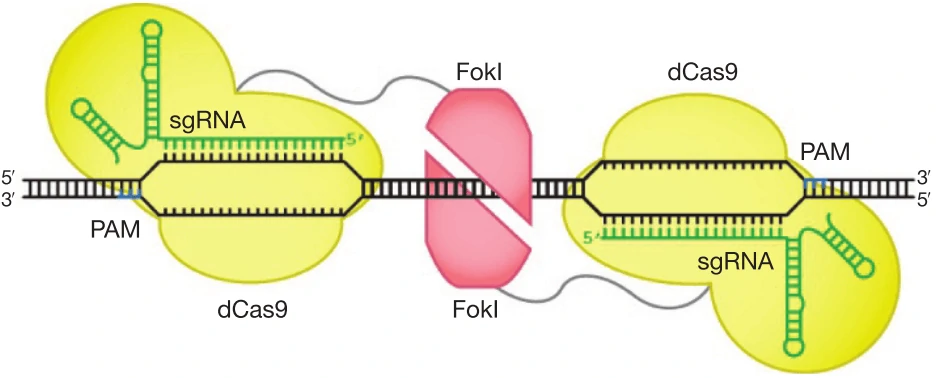
f. Improve DNA cleavage specificity using fusions of dCas9 with FokI nuclease domain (fCas9) (Figure 6) [84,85].